Les Informations Présentées Sur Ce Site Ne Constituent Pas Un Avis Médical. Nous Ne Vendons Rien. L'Exactitude De La Traduction N'Est Pas Garantie. Clause De Non-Responsabilité
Antipsories, systémiqueHumira
Résumé
Qu'est-ce que Humira?
Humira (adalimumab) est une protéine injectable (anticorps) utilisée pour traiter polyarthrite rhumatoïde juvénile idiopathe arthrite arthrite psoriatique spondylarthrite ankylosante et psoriasis en plaques . Humira est également utilisé pour traiter la maladie de Crohn après que d'autres médicaments ont été essayés sans traitement réussi des symptômes.
Quels sont les effets secondaires de Humira?
Les effets secondaires courants de Humira incluent
- Réactions du site d'injection (rougeur démangeaisons douleurs douloureux ou saignements)
- mal de tête
- nez de nez
- sinus douleur ou
- Douleurs d'estomac.
Dites à votre médecin si vous avez des effets secondaires graves de Humira, notamment:
- rythme cardiaque rapide / irrégulier / martelant
- douleurs à l'estomac
- sang dans les tabourets
- changements mentaux / d'humeur
- maux de tête sévères
- ecchymoses ou saignements faciles
- urine sombre
- Les yeux et la peau jaunissent
- Douleur ou gonflement des jambes
- engourdissement ou picotement des bras / mains / jambes / pieds
- déséquilibre
- faiblesse musculaire inexpliquée
- difficulté de parler / mâcher / avaler / mouvements faciaux
- changements de vision
- fatigue extrême
- douleur articulaire ou
- Éruption cutanée en forme de papillon sur le nez et les joues.
Dosage pour Humira
La dose recommandée de Humira pour les patients adultes atteints de polyarthrite rhumatoïde (RA) arthrite psoriatique (PSA) ou ankylosing spondylite (AS) est administré à 40 mg toutes les deux semaines. Le dosage pédiatrique est déterminé par le poids de l'enfant.
Quelles substances ou suppléments de médicaments interagissent avec Humira?
D'autres médicaments peuvent interagir avec Humira. Dites à votre médecin tous les médicaments et suppléments en vente libre et en vente libre que vous utilisez.
Humira During Grossesse et Breastfeeding
Pendant la grossesse, Humira ne doit être utilisé que lorsqu'il est prescrit. On ne sait pas si ce médicament passe dans le lait maternel. Des médicaments similaires passent dans le lait maternel. Consultez votre médecin avant l'allaitement.
Informations Complémentaires
Notre centre de médicaments à effets secondaires de Humira (Adalimumab) offre une vue complète des informations sur les médicaments disponibles sur les effets secondaires potentiels lors de la prise de ce médicament.
Informations sur les médicaments de la FDA
- Description de la drogue
- Indications
- Dosage
- Effets secondaires
- Interactions médicamenteuses
- Avertissements
- Surdosage
- Pharmacologie clinique
- Guide des médicaments
AVERTISSEMENT
Infections graves et tumeurs malignes
Infections graves
Les patients traités par Humira courent un risque accru de développer des infections graves qui peuvent entraîner une hospitalisation ou une mort [voir avertissements et PRÉCAUTIONS ]. La plupart des patients qui ont développé ces infections prenaient des immunosuppresseurs concomitants tels que le méthotrexate ou les corticostéroïdes.
Arrêtez Humira si un patient développe une infection grave ou une septicémie.
Les infections signalées comprennent:
- Tuberculose active (TB) comprenant la réactivation de la tuberculose latente. Les patients atteints de tuberculose ont fréquemment présenté une maladie disséminée ou extrapulmonaire. Tester les patients pour la tuberculose latente avant l'utilisation de Humira et pendant le traitement. Initier un traitement pour la tuberculose latente avant l'utilisation de Humira.
- Infections fongiques invasives, y compris l'histoplasmose, la coccidioïdomycose candidose aspergillose blastomycose et pneumocystose. Les patients atteints d'histoplasmose ou d'autres infections fongiques invasives peuvent présenter une maladie disséminée plutôt que localisée. Les tests d'antigène et d'anticorps pour l'histoplasmose peuvent être négatifs chez certains patients atteints d'une infection active. Considérons le traitement antifongique empirique chez les patients à risque d'infections fongiques invasives qui développent une maladie systémique grave.
- Infections virales et autres bactériennes dues à des agents pathogènes opportunistes, notamment Legionella et Listeria.
Considérez soigneusement les risques et les avantages du traitement avec Humira avant de lancer une thérapie chez les patients infectés chroniques ou récurrentes.
Surveillez les patients étroitement pour le développement de signes et symptômes d'infection pendant et après le traitement par Humira, y compris le développement possible de la tuberculose chez les patients qui ont testé négatif pour une infection à la tuberculose latente avant de lancer un traitement [voir avertissements et PRÉCAUTIONS et Effets indésirables ].
Malignité
Lymphome et autres tumeurs malignes. PRÉCAUTIONS ]. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL) a rare type of T-cell lymphoma have been reported in patients treated with TNF blockers including . These cases have had a very aggressive disease course et have been fatal. The majority of reported TNF blocker cases have occurrouge in patients with Crohn's disease or ulcerative colitis et the majority were in adolescent et young adult males. Almost all these patients had received treatment with azathioprine or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Description de Humira
Humira (adalimumab) est un anticorps monoclonal IgG1 humain recombinant spécifique du facteur de nécrose tumorale humaine (TNF). Humira a été créé en utilisant la technologie des phages d'affichage, ce qui a résulté en un anticorps avec des régions variables de chaîne lourde et légère dérivées humaines et des régions constantes d'IgG1 humaines: k. L'adalimumab est produit par la technologie d'ADN recombinant dans un système d'expression de cellules mammifères et est purifié par un processus qui comprend des étapes d'inactivation virale et d'élimination spécifiques. Il se compose de 1330 acides aminés et a un poids moléculaire d'environ 148 kilodalts.
Humira est fourni comme une solution stérile sans conservateur d'adalimumab pour l'administration sous-cutanée. Le produit médicamenteux est fourni comme un stylo préfabillé à usage unique (stylo Humira) en tant que seringue en verre préfabillée de 1 ml à usage unique ou comme flacon à usage institutionnel à usage unique. Closed dans le stylo se trouve une seringue en verre préfabillée de 1 ml à usage unique. La solution de Humira est claire et incolore avec un pH d'environ 5,2.
Chaque seringue préfabillée de 80 mg / 0,8 ml ou un stylo préfabillé offre 0,8 ml (80 mg) de produit médicamenteux. Chaque 0,8 ml de Humira contient de l'adalimumab (80 mg) de mannitol (NULL,6 mg) en polysorbate 80 (NULL,8 mg) et de l'eau pour l'injection USP.
Chaque seringue préfabillée de 40 mg / 0,4 ml ou un stylo préfabillé offre 0,4 ml (40 mg) de produit médicamenteux. Chaque 0,4 ml de Humira contient de l'adalimumab (40 mg) de mannitol (NULL,8 mg) en polysorbate 80 (NULL,4 mg) et de l'eau pour l'injection USP.
Chaque 40 mg / 0,8 ml de la seringue prérempliée ou du flacon à usage institutionnel à usage unique délivre 0,8 ml (40 mg) de produit médicamenteux. Each 0.8 mL of HUMIRA contains adalimumab (40 mg) citric acid monohydrate (1.04 mg) dibasic sodium phosphate dihydrate (1.22 mg) mannitol (9.6 mg) monobasic sodium phosphate dihydrate (0.69 mg) polysorbate 80 (0.8 mg) sodium chloride (4.93 mg) sodium citrate (NULL,24 mg) et l'eau pour l'injection USP. L'hydroxyde de sodium est ajouté si nécessaire pour régler le pH.
Chaque seringue préfabillée de 20 mg / 0,2 ml offre 0,2 ml (20 mg) de produit médicamenteux. Chaque 0,2 ml de Humira contient de l'adalimumab (20 mg) de mannitol (NULL,4 mg) en polysorbate 80 (NULL,2 mg) et de l'eau pour l'injection USP.
Chaque seringue préfabillée de 20 mg / 0,4 ml offre 0,4 ml (20 mg) de produit médicamenteux. Each 0.4 mL of HUMIRA contains adalimumab (20 mg) citric acid monohydrate (0.52 mg) dibasic sodium phosphate dihydrate (0.61 mg) mannitol (4.8 mg) monobasic sodium phosphate dihydrate (0.34 mg) polysorbate 80 (0.4 mg) sodium chloride (2.47 mg) sodium citrate (NULL,12 mg) et eau pour l'injection USP. L'hydroxyde de sodium est ajouté si nécessaire pour régler le pH.
Chaque seringue préfabillée de 10 mg / 0,1 ml offre 0,1 ml (10 mg) de produit médicamenteux. Chaque 0,1 ml de Humira contient de l'adalimumab (10 mg) de mannitol (NULL,2 mg) en polysorbate 80 (NULL,1 mg) et de l'eau pour l'injection USP.
Chaque seringue préfabillée de 10 mg / 0,2 ml offre 0,2 ml (10 mg) de produit médicamenteux. Each 0.2 mL of HUMIRA contains adalimumab (10 mg) citric acid monohydrate (0.26 mg) dibasic sodium phosphate dihydrate (0.31 mg) mannitol (2.4 mg) monobasic sodium phosphate dihydrate (0.17 mg) polysorbate 80 (0.2 mg) sodium chloride (1.23 mg) sodium citrate (NULL,06 mg) et l'eau pour l'injection USP. L'hydroxyde de sodium est ajouté si nécessaire pour régler le pH.
Utilisations pour Humira
Polyarthrite rhumatoïde
Humira est indiqué pour réduire les signes et symptômes induisant une réponse clinique majeure inhibant la progression des dommages structurels et améliorant polyarthrite rhumatoïde . Humira peut être utilisé seul ou en combinaison avec du méthotrexate ou d'autres médicaments anti-rhumatismaux modifiant les maladies non biologiques (DMARD).
Arthrite idiopathique juvénile
Humira est indiqué pour réduire les signes et symptômes d'une arthrite idiopathique juvénile modérément à gravement active chez les patients de 2 ans et plus. Humira peut être utilisé seul ou en combinaison avec le méthotrexate.
Arthrite psoriatique
Humira est indiqué pour réduire les signes et symptômes inhibant la progression des dommages structurels et améliorant la fonction physique chez les patients adultes atteints d'arthrite psoriasique active. Humira peut être utilisé seul ou en combinaison avec des ARM non biologiques.
Spondylarthrite ankylosante
Humira est indiqué pour réduire les signes et symptômes chez les patients adultes atteints de spondylarthrite ankylosante active.
Maladie de Crohn
Humira est indiqué pour le traitement de la maladie de Crohn modérément à gravement active chez les adultes et les patients pédiatriques de 6 ans et plus.
Rectocolite hémorragique
Humira est indiqué pour le traitement de la colite ulcéreuse modérément à sévère chez les adultes et les patients pédiatriques de 5 ans et plus.
Limitations d'utilisation
L'efficacité de Humira n'a pas été établie chez les patients qui ont perdu une réponse ou étaient intolérants aux bloqueurs TNF [voir Études cliniques ].
Psoriasis en plaques
Humira est indiqué pour le traitement des patients adultes atteints de psoriasis de plaque chronique modéré à sévère qui sont candidats à une thérapie systémique ou à la photothérapie et lorsque d'autres thérapies systémiques sont médicalement moins appropriées. Humira ne doit être administré qu'aux patients qui seront étroitement surveillés et auront des visites de suivi régulières avec un médecin [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Hidradénite suppurative
Humira est indiqué pour le traitement de la Hidradénite suppurativa modérée à sévère chez les patients âgés de 12 ans et plus.
Uvéite
Humira est indiqué pour le traitement de l'intermédiaire non infectieux et de la panuvéite chez les adultes et les patients pédiatriques de 2 ans et plus.
Dosage pour Humira
Polyarthrite rhumatoïde Arthrite psoriatique And Spondylarthrite ankylosante
La dose sous-cutanée recommandée de Humira pour les patients adultes atteints de polyarthrite rhumatoïde (RA) arthrite psoriatique (PSA) ou spondylarthrite ankylosante (AS) est administrée de 40 mg toutes les deux semaines. Méthotrexate (MTX) D'autres glucocorticoïdes non biologiques des Glucocorticoïdes non stéroïdiens (AINS) et / ou analgésiques peuvent être poursuivis pendant le traitement avec Humira. Dans le traitement de la PR, certains patients ne prenant pas de MTX concomitants peuvent tirer un avantage supplémentaire de l'augmentation du dosage de Humira à 40 mg chaque semaine ou 80 mg toutes les deux semaines.
Arthrite idiopathique juvénile Or Uvéite pédiatrique
La dose sous-cutanée recommandée de Humira pour les patients de 2 ans et plus avec une arthrite idiopathique juvénile polyarticulaire (JIA) ou une uvéite pédiatrique est basée sur le poids comme indiqué ci-dessous. Les glucocorticoïdes MTX et / ou les analgésiques peuvent être poursuivis pendant le traitement avec Humira.
| Poids pédiatrique (2 ans et plus) | Dosage recommandé |
| 10 kg (22 lb) à moins de 15 kg (33 lb) | 10 mg toutes les deux semaines |
| 15 kg (33 lb) à moins de 30 kg (66 lb) | 20 mg toutes les deux semaines |
| 30 kg (66 lb) et plus | 40 mg toutes les deux semaines |
Humira n'a pas été étudié chez des patients atteints de JIA polyarticulaire ou d'uvéite pédiatrique de moins de 2 ans ou chez les patients avec un poids inférieur à 10 kg.
Maladie de Crohn
Adultes
Le dosage sous-cutané recommandé de Humira pour les patients adultes atteints de la maladie de Crohn (CD) est de 160 mg initialement le jour 1 (donné en une journée ou divisé sur deux jours consécutifs) suivi de 80 mg deux semaines plus tard (jour 15). Deux semaines plus tard (jour 29) commencent une dose de 40 mg toutes les deux semaines. Les aminosalicylates et / ou les corticostéroïdes peuvent être poursuivis pendant le traitement avec Humira. Azathioprine 6-mercaptopurine (6-MP) [voir AVERTISSEMENTS AND PRÉCAUTIONS ] ou MTX peut être poursuivi pendant le traitement avec Humira si nécessaire.
Pédiatrie
La dose sous-cutanée recommandée de Humira pour les patients pédiatriques de 6 ans et plus avec la maladie de Crohn (CD) est basée sur le poids corporel comme indiqué ci-dessous:
| Poids pédiatrique | Dosage recommandé | |
| Jours 1 à 15 | À partir du jour 29 | |
| 17 kg (37 lb) à moins de 40 kg (88 lb) | Jour 1: 80 mg Jour 15: 40 mg | 20 mg toutes les deux semaines |
| 40 kg (88 lb) et plus | Jour 1: 160 mg (dose unique ou divisé sur deux jours consécutifs) Jour 15: 80 mg | 40 mg toutes les deux semaines |
Rectocolite hémorragique
Adultes
Le dosage sous-cutané recommandé de Humira pour les patients adultes atteints de colite ulcéreuse est de 160 mg initialement le jour 1 (donné en une journée ou divisé sur deux jours consécutifs) suivi de 80 mg deux semaines plus tard (jour 15). Deux semaines plus tard (jour 29), continuent avec une dose de 40 mg toutes les deux semaines.
Arrêtez Humira chez les patients adultes sans signe de rémission clinique de huit semaines (jour 57) du traitement. Les aminosalicylates et / ou les corticostéroïdes peuvent être poursuivis pendant le traitement avec Humira. Azathioprine et 6-mercaptopurine (6-MP) [voir AVERTISSEMENTS AND PRÉCAUTIONS ] peut être poursuivi pendant le traitement avec Humira si nécessaire.
Pédiatrie
La dose sous-cutanée recommandée de Humira pour les patients pédiatriques de 5 ans et plus avec une colite ulcéreuse est basée sur le poids corporel comme indiqué ci-dessous:
| Poids pédiatrique | Dosage recommandé | |
| Jours 1 à 15 | À partir du jour 29* | |
| 20 kg (44 lb) à moins de 40 kg (88 lb) | Jour 1: 80 mg Jour 8: 40 mg Jour 15: 40 mg | 40 mg toutes les deux semaines or 20 mg every week |
| 40 kg (88 lb) et plus | Jour 1: 160 mg (dose unique ou divisé sur deux jours consécutifs) Jour 8: 80 mg Jour 15: 80 mg | 80 mg toutes les deux semaines ou 40 mg chaque semaine |
| * Continuez la dose pédiatrique recommandée chez les patients qui ont 18 ans et qui sont bien contrôlés sur leur régime Humira. |
Psoriasis en plaques Or Uvéite adulte
La dose sous-cutanée recommandée de Humira pour les patients adultes atteints de psoriasis en plaque (PS) ou d'uvéite (UV) est une dose initiale de 80 mg suivie de 40 mg étant donné toutes les deux semaines à partir d'une semaine après la dose initiale. L'utilisation de Humira dans le PS chronique modéré à sévère au-delà d'un an n'a pas été évaluée dans des études cliniques contrôlées.
Hidradénite suppurative
Adultes
La dose sous-cutanée recommandée de Humira pour les patients adultes atteints de Hidradénite suppurativa (HS) est une dose initiale de 160 mg (donnée en une journée ou divisée sur deux jours consécutifs) suivi de 80 mg deux semaines plus tard (jour 15). Commencez 40 mg chaque semaine ou 80 mg toutes les deux semaines pour doser deux semaines plus tard (jour 29).
Adolescents
La dose sous-cutanée recommandée de Humira pour les adolescents de 12 ans et plus pesant au moins 30 kg avec une Hidradénite suppurativa (HS) est basée sur le poids corporel comme indiqué ci-dessous [voir Utiliser dans des populations spécifiques et Pharmacologie clinique ]::
| Poids corporel des adolescents (12 ans et plus) | Dosage recommandé |
| 30 kg (66 lb) à moins de 60 kg (132 lb) |
|
| 60 kg (132 lb) et plus |
|
Surveillance pour évaluer la sécurité
Avant de lancer Humira et périodiquement pendant la thérapie, évaluez les patients pour tuberculose et test for latent infection [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Considérations générales pour l'administration
Humira est destiné à être utilisé sous la direction et la supervision d'un médecin. Un patient peut s'auto-injecter Humira ou un soignant peut injecter Humira en utilisant le stylo Humira ou une seringue préfilée si un médecin détermine qu'il est approprié et avec un suivi médical si nécessaire après une formation appropriée dans la technique d'injection sous-cutanée.
Humira peut être retiré du réfrigérateur pendant 15 à 30 minutes avant d'injecter pour permettre au liquide de venir à température ambiante. Ne retirez pas le capuchon ou le couvercle tout en lui permettant d'atteindre la température ambiante. Inspectez soigneusement la solution dans la seringue préreffilée du stylo Humira ou le flacon d'institution à dose unique pour les particules et la décoloration avant l'administration sous-cutanée. Si les particules et les décolorations sont notées, n'utilisez pas le produit. Humira ne contient pas de conservateurs; Par conséquent, jetez des parties inutilisées de médicaments restant à partir de la seringue. Remarque: Instruisez les patients sensibles au latex pour ne pas manipuler la couverture à l'aiguille de l'humira 40 mg / 0,8 ml de stylo et 40 mg / 0,8 ml 20 mg / 0,4 ml et 10 mg / 0,2 ml de seringue préfabillée car elle peut contenir le latex en caoutchouc naturel [voir Comment fourni / Stockage et manipulation ].
Instruisez les patients utilisant le stylo Humira ou la seringue préfilée pour injecter la quantité totale dans la seringue en fonction des instructions fournies dans les instructions d'utilisation [voir Instructions pour une utilisation ].
Des injections doivent se produire sur des sites séparés de la cuisse ou de l'abdomen. Faites tourner les sites d'injection et ne donnez pas d'injections dans des zones où la peau est soumis à tendre rouge ou dur. »
Le flacon à usage institutionnel à dose Humira est pour l'administration dans un cadre institutionnel uniquement comme un bureau ou une clinique d'un médecin hospitalier. Retirez la dose à l'aide d'une aiguille et d'une seringue stériles et administrez rapidement par un professionnel de la santé dans un cadre institutionnel. Administrer une seule dose par flacon. Le flacon ne contient pas de conservateurs; donc éliminer les parties inutilisées.
Comment fourni
Dosage Forms And Strengths
est une solution claire et incolore disponible comme:
- Pen (Humira Pen)
- Injection: 80 mg / 0,8 ml dans un stylo à dose unique.
- Injection: 40 mg / 0,8 ml dans un stylo à dose unique.
- Injection: 40 mg / 0,4 ml dans un stylo à dose unique.
- Seringue prérempli
- Injection: 80 mg / 0,8 ml dans une seringue en verre préreadis à dose.
- Injection: 40 mg / 0,8 ml dans une seringue en verre préremplée à dose unique.
- Injection: 40 mg / 0,4 ml dans une seringue en verre préreffilée à dose unique.
- Injection: 20 mg / 0,4 ml dans une seringue en verre préreffilée à dose unique.
- Injection: 20 mg / 0,2 ml dans une seringue en verre préreadis à dose.
- Injection: 10 mg / 0,2 ml dans une seringue en verre préreadis à dose.
- Injection: 10 mg / 0,1 ml dans une seringue en verre préreffilée à dose unique.
- Vial à usage institutionnel à dose unique
- Injection: 40 mg / 0,8 ml dans un flacon de verre à dose unique pour un usage institutionnel uniquement.
Stockage et manipulation
® (adalimumab) est fourni en tant que solution claire stérile et incolore sans conservateur pour l'administration sous-cutanée. Les configurations d'emballage suivantes sont disponibles.
- Pen Carton - 40 mg/0.8 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-4339-02.
- Pen Carton - 40 mg / 0,4 ml
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0554-02.
- Pen Carton †80 mg/0.8 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0124-02.
- Pen 40 mg/0.8 mL - Starter Package for Crohn's Disease Rectocolite hémorragique or Hidradénite suppurative
- is supplied in a carton containing 6 alcohol preps et 6 dose trays (Starter Package for Maladie de Crohn Rectocolite hémorragique or Hidradénite suppurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-4339-06.
- Pen 40 mg / 0,4 ml - Starter Package for Crohn's Disease Rectocolite hémorragique or Hidradénite suppurative
- is supplied in a carton containing 6 alcohol preps et 6 dose trays (Starter Package for Maladie de Crohn Rectocolite hémorragique or Hidradénite suppurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0554-06.
- Pen 80 mg/0.8 mL - Starter Package for Crohn's Disease Rectocolite hémorragique or Hidradénite suppurative
- is supplied in a carton containing 4 alcohol preps et 3 dose trays (Starter Package for Maladie de Crohn Rectocolite hémorragique or Hidradénite suppurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0124-03.
- Pen 40 mg/0.8 mL - Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package
- is supplied in a carton containing 4 alcohol preps et 4 dose trays (Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-4339-07.
- Pen 40 mg / 0,4 ml - Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package
- is supplied in a carton containing 4 alcohol preps et 4 dose trays (Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0554-04.
- Pen 80 mg/0.8 mL et 40 mg / 0,4 ml - Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package
- is supplied in a carton containing 4 alcohol preps et 3 dose trays (Psoriasis Uvéite or Adolescent Hidradénite suppurative Starter Package). One dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The other two dose trays each consist of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-1539-03.
- Pen 80 mg/0.8 mL †Starter Package for Colite ulcéreuse pédiatrique (4 count)
- is supplied in a carton containing 4 alcohol preps et 4 dose trays (Starter Package for Colite ulcéreuse pédiatrique). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0124-04.
- Seringue prérempli Carton - 40 mg/0.8 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-3799-02.
- Seringue prérempli Carton - 40 mg / 0,4 ml
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0243-02.
- Seringue prérempli Carton - 20 mg/0.4 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 20 mg/0.4 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-9374-02.
- Seringue prérempli Carton - 20 mg/0.2 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 20 mg/0.2 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0616-02.
- Seringue prérempli Carton - 10 mg/0.2 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 10 mg/0.2 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-6347-02.
- Seringue prérempli Carton - 10 mg/0.1 mL
- is supplied in a carton containing two alcohol preps et two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 10 mg/0.1 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0817-02.
- Seringue prérempli 40 mg/0.8 mL - Maladie pédiatrique de Crohn Starter Package (6 count)
- is supplied in a carton containing 6 alcohol preps et 6 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-3799-06.
- Seringue prérempli 80 mg/0.8 mL - Maladie pédiatrique de Crohn Starter Package (3 count)
- is supplied in a carton containing 4 alcohol preps et 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-2540-03.
- Seringue prérempli 40 mg/0.8 mL - Maladie pédiatrique de Crohn Starter Package (3 count)
- is supplied in a carton containing 4 alcohol preps et 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of . The needle cover may contain natural frotterber latex. The NDC Le numéro est 0074-3799-03.
- Seringue prérempli 80 mg/0.8 mL et 40 mg / 0,4 ml - Maladie pédiatrique de Crohn Starter Package (2 count)
- is supplied in a carton containing 2 alcohol preps et 2 dose trays (Pediatric Starter Package). One dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of . The other dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg / 0,4 ml of . The black needle cover is pas made with natural frotterber latex. The NDC Le numéro est 0074-0067-02.
- Vial à usage institutionnel à dose unique Carton - 40 mg/0.8 mL
- is supplied for institutional use only in a carton containing a single-dose glass vial providing 40 mg/0.8 mL of . The vial stopper is pas made with natural frotterber latex. The NDC Le numéro est 0074-3797-01.
Stockage et stabilité
N'utilisez pas au-delà de la date d'expiration du conteneur. Humira doit être réfrigéré à 36 ° F à 46 ° F (2 ° C à 8 ° C). Ne congelez pas. N'utilisez pas si gelé même s'il a été décongelé.
Stockez en carton d'origine jusqu'à l'heure de l'administration pour se protéger de la lumière.
Si nécessaire, par exemple, lorsque vous déplacez, Humira peut être stocké à température ambiante jusqu'à un maximum de 77 ° F (25 ° C) pendant une période pouvant aller jusqu'à 14 jours avec une protection contre la lumière. Humira doit être jeté s'il n'est pas utilisé dans les 14 jours. Enregistrez la date où Humira est enlevé pour la première fois du réfrigérateur dans les espaces fournis sur le carton et le plateau à dose.
Ne stockez pas Humira dans une chaleur ou un froid extrême.
Abbvie Inc. North Chicago IL 60064 U.S.A. Numéro de licence américaine. Révisé: février 2021
Effets secondaires for Humira
Les effets indésirables cliniquement significatifs suivants sont décrits ailleurs dans l'étiquetage:
- Infections graves [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Tumeurs malignes [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Réactions d'hypersensibilité [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Réactivation du virus de l'hépatite B [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Réactions neurologiques [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Réactions hématologiques [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Insuffisance cardiaque [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
- Auto-immunité [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
Expérience des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions de réaction indésirables très variables observées dans les essais cliniques d'un médicament ne peuvent pas être directement comparées aux taux dans les essais cliniques d'un autre médicament et ne peuvent pas refléter les taux observés dans la pratique.
La réaction indésirable la plus courante avec Humira a été les réactions du site d'injection. Dans les essais de placebocallé, 20% des patients traités par Humira ont développé des réactions de sites d'injection (érythème et / ou douleur ou gonflement de l'hémorragie qui démange) par rapport à 14% des patients recevant un placebo. La plupart des réactions du site d'injection ont été décrites comme légères et ne nécessitaient généralement pas l'arrêt du médicament.
La proportion de patients qui ont interrompu le traitement en raison des effets indésirables pendant la partie contrôlée par placebo en double aveugle chez les patients atteints de PR (c'est-à-dire des études RA-I RAII RA-III et RA-IV) était de 7% pour les patients prenant Humira et 4% pour les patients traités par placebo. Les effets indésirables les plus courants conduisant à l'arrêt de l'humira dans ces études de PR étaient la réaction clinique (NULL,7%).
Infections
Dans les parties contrôlées des 39 essais cliniques Global Humira chez les patients adultes atteints de PSA de PSA comme CD UC PS HS et UV, le taux d'infections graves était de 4,3 pour 100 années de patient chez 7973 patients traités par Humira contre un taux de 2,9 pour 100 années-patients chez 4848 patients traités par contrôle. Infections graves observées comprenant l'arthrite septique de la pneumonie prothétique et postsurgical infections erysipelas cellulitis diverticulite et pyelonephritis [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Tuberculose et infections opportunistes
Dans 52 essais cliniques contrôlés et incontrôlés globaux en PSA RA en tant que CD UC PS HS et UV, qui comprenait 24605 patients traités par Humira, le taux de tuberculose actif signalé était de 0,20 pour 100 années de patient et le taux de conversion PPD positive était de 0,09 pour 100 patients-et-patients. Dans un sous-groupe de 10113 patients traités aux États-Unis et au Canada Humira, le taux de tuberculose actif signalé était de 0,05 pour 100 années de patient et le taux de conversion PPD positive était de 0,07 pour 100 années de patient. Ces essais comprenaient des rapports de TB péritonéale lymphatique et pulmonaire miliaire. AVERTISSEMENTS AND PRÉCAUTIONS ].
Auto-anticorps
Dans la polyarthrite rhumatoïde, les essais ont contrôlé 12% des patients traités par Humira et 7% des patients traités par placebo qui avaient des titres ANA de base négatifs ont développé des titres positifs à la semaine 24. Deux patients sur 3046 traités par Humira ont développé des signes cliniques suggérant un nouveau recentrage lupus - syndrome de type. Les patients se sont améliorés après l'arrêt du traitement. Aucun patient n'a développé une néphrite de lupus ou des symptômes du système nerveux central. L'impact du traitement à long terme avec Humira sur le développement de maladies auto-immunes est inconnu.
Élévations enzymatiques du foie
Il y a eu des rapports de réactions hépatiques graves, notamment une insuffisance hépatique aiguë chez les patients recevant des bloqueurs TNF. Dans les essais contrôlés de phase 3 de Humira (40 mg de SC toutes les deux semaines) chez les patients atteints de PSA de PR et comme avec une durée de la période de contrôle allant de 4 à 104 semaines Alt Alt ≥ 3 x Uln s'est produit chez 3,5% des patients traités à Humira et 1,5% des patients contrôlés par contrôle. Étant donné que bon nombre de ces patients dans ces essais prenaient également des médicaments qui provoquaient des élévations enzymatiques du foie (par exemple les AINS MTX), la relation entre Humira et les élévations enzymatiques du foie n'est pas claire. Dans un essai contrôlé de phase 3 de Humira chez les patients atteints de JIA polyarticulaire qui était de 4 à 17 ans, des altitudes alt ≥ 3 x ULN se sont produites chez 4,4% des patients traités à Humira et 1,5% des patients traités par le contrôle (ALT plus commun que l'AST); Les élévations du test des enzymes hépatiques étaient plus fréquentes chez les personnes traitées avec la combinaison de Humira et MTX que celles traitées avec Humira seul. En général, ces élévations n'ont pas conduit à l'arrêt du traitement Humira. Aucune altitude alt ≥ 3 x uln ne s'est produite dans l'étude ouverte de Humira chez les patients atteints de JIA polyarticulaire qui étaient 2 à <4 years.
Dans les essais contrôlés de phase 3 de Humira (doses initiales de 160 mg et 80 mg ou 80 mg et 40 mg aux jours 1 et 15 respectivement suivis de 40 mg toutes les deux semaines) chez des patients adultes atteints de la maladie de Crohn avec une durée de période de contrôle allant de 4 à 52 semaines Alt Else ≥ 3 x ULN ont eu lieu chez 0,9% des patients traités par Humira et 0,9% de patients traités par le contrôle. Dans l'essai de phase 3 de Humira chez les patients pédiatriques atteints de la maladie de Crohn, qui a évalué l'efficacité et l'innocuité de deux schémas de dose d'entretien basés sur le poids corporel après une thérapie d'induction basée sur le poids corporel jusqu'à 52 semaines de traitement ALT Elose ≥ 3 x Uln se sont produits dans 2,6% (5/192) de patients dont 4 ont reçu des immunosuppressants concomitants au niveau de la base de la base; Aucun de ces patients ne s'est arrêté en raison d'anomalies dans les tests ALT. Dans la phase contrôlée 3, les essais de Humira (doses initiales de 160 mg et 80 mg aux jours 1 et 15 respectivement suivis de 40 mg toutes les deux semaines) chez des patients adultes atteints de CU avec une durée de période de contrôle allant de 1 à 52 semaines Alt Elose ≥3 x ULN chez 1,5% de patients traités à Humira et 1,0% des patients traités par contrôle. In the controlled Phase 3 trial of HUMIRA in patients with pediatric ulcerative colitis (N=93) which evaluated efficacy and safety of a maintenance dose of 0.6 mg/kg (maximum of 40 mg) every other week (N=31) and a maintenance dose of 0.6 mg/kg (maximum of 40 mg) every week (N=32) following body weight based induction doses of 2.4 mg/kg (maximum of 160 mg) à la semaine 0 et la semaine 1 et 1,2 mg / kg (maximum de 80 mg) à la semaine 2 (n = 63) ou une dose d'induction de 2,4 mg / kg (maximum de 160 mg) à la semaine 0 placebo à la semaine 1 et 1,2 mg / kg (maximum de 80 mg) à la semaine 0 à la semaine 1 et 1,2 mg / kg (maximum de 80 mg) à la semaine 2 (N = 30) Eléments alt ≥ 3 x Uln s'est produit dans 1,1% (1) des patients. Dans les essais contrôlés de phase 3 de Humira (dose initiale de 80 mg puis 40 mg toutes les deux semaines) chez les patients atteints de PS avec une durée de période de contrôle allant de 12 à 24 semaines Alt Alt ≥ 3 x ULN s'est produit chez 1,8% des patients traités à Humira et 1,8% des patients traités par contrôle. Dans des essais contrôlés de Humira (doses initiales de 160 mg à la semaine 0 et 80 mg à la semaine 2 suivie de 40 mg chaque semaine à partir de la semaine 4) dans des sujets avec HS avec une durée de période de contrôle allant de 12 à 16 semaines Alt Elose ≥ 3 x ULN sur les sujets traités à 0,3% de Humira et 0,6% des sujets traités par contrôle. Dans les essais contrôlés de Humira (doses initiales de 80 mg à la semaine 0 suivies de 40 mg toutes les deux semaines à partir de la semaine 1) chez des patients adultes atteints d'uvéite avec une exposition de 165,4 PYS et 119,8 PYS chez les patients traités par Humira et traités par le contrôle respectivement ≥ 3 x ULN ont eu lieu chez les patients traités par Humira et 2,4% de patients traités par Humira.
Autres réactions indésirables
Polyarthrite rhumatoïde Études cliniques
Les données décrites ci-dessous reflètent l'exposition à Humira chez 2468 patients, dont 2073 exposés pendant 6 mois 1497 exposés pendant plus d'un an et 1380 dans des études adéquates et bien contrôlées (études RA-I RA-II RA-III et RA-IV). Humira a été étudié principalement dans les essais sous-contrôlés de placement et dans des études de suivi à long terme pour une durée maximale de 36 mois. La population avait un âge moyen de 54 ans, 77% étaient des femmes 91% étaient du Caucasien et avaient une polyarthrite rhumatoïde modérément à sévèrement active. La plupart des patients ont reçu 40 mg Humira toutes les deux semaines [voir Études cliniques ].
Le tableau 1 résume les réactions rapportées à un taux d'au moins 5% chez les patients traités avec Humira 40 mg toutes les deux semaines par rapport au placebo et avec une incidence supérieure au placebo. Dans l'étude RA-III, les types et les fréquences des effets indésirables au cours de l'extension ouverte de deuxième année étaient similaires à ceux observés dans la partie en double aveugle d'un an.
Tableau 1: Réactions indésirables rapportées par ≥ 5% des patients traités par Humira pendant la période contrôlée par placebo d'études RA regroupées (études RA-I RA-II RA-III et RA-IV)
| 40 mg subcutaneous Every Autre Week (N = 705) | Placebo (N = 690) | |
| Réaction indésirable (terme préféré) | ||
| Respiratoire | ||
| Infection respiratoire supérieure | 17% | 13% |
| Sinusite | 11% | 9% |
| Syndrome de la grippe | 7% | 6% |
| Gastro-intestinal | ||
| Nausée | 9% | 8% |
| Douleurs abdominales | 7% | 4% |
| Tests de laboratoire * | ||
| Test de laboratoire anormal | 8% | 7% |
| Hypercholestérolémie | 6% | 4% |
| Hyperlipidémie | 7% | 5% |
| Hématurie | 5% | 4% |
| La phosphatase alcaline a augmenté | 5% | 3% |
| Autre | ||
| Mal de tête | 12% | 8% |
| Éruption cutanée | 12% | 6% |
| Blessure accidentelle | 10% | 8% |
| Réaction du site d'injection ** | 8% | 1% |
| Maux de dos | 6% | 4% |
| Infection des voies urinaires | 8% | 5% |
| Hypertension | 5% | 3% |
| * Des anomalies d'essai en laboratoire ont été signalées comme des réactions indésirables dans les essais européens ** n'inclut pas le site d'injection érythème démangeaisons l'hémorragie douleur ou gonflement |
Réactions indésirables moins courantes dans les études cliniques de la polyarthrite rhumatoïde
Autre infrequent serious adverse reactions that ne pas appear in the Warnings et Precautions or Adverse Reaction sections that occurrouge at an incidence of less than 5% in -treated patients in RA studies were:
Corps dans son ensemble: Douleur en extrémité chirurgie de la douleur pelvienne Torax Pain
Système cardiovasculaire: Arythmie Fibrillation auriculaire Douleur thoracique Trouble coronarien Trouble cardiaque Hypertensive Encéphalopathie Myocarde Palpitation Péricarde péricardique Péricardite Syncope Tachycardie
Système digestif: Cholécystite cholelitase œsophagite gastroentérite gastro-intestinal hémorragie nécrose hépatique vomissements
Système endocrinien: Trouble parathyroïdien
Système hémic et lymphatique: Agranulocytose Polycythémie
Troubles métaboliques et nutritionnels: Déshydratation guérison cétose anormale paraprotéinémie périphérique œdème
Système musculo-squelettique: Arthrite Trouble osseux Fracture osseuse (non spontanée) Nécrose osseuse Trouble articulaire Muscle Crampes
Néoplasie: Adénome
Système nerveux: Confusion paresthésie hématome sous-dural tremblement
Respiratoire System: Bronchospasme de l'asthme La fonction pulmonaire de la dyspnée a diminué l'épanchement pleural
Sens spéciaux: Cataracte
Thrombose: Jambe de thrombose
Système urogénital: Trouble menstruel du calcul rénal de la cystite
Arthrite idiopathique juvénile Études cliniques
En général, les effets indésirables des passants traités à Humira dans les essais d'arthrite idioparthique juvénile polyarticulaire (JIA) (études Jia-I et Jia-II) [voir Études cliniques ] étaient similaires en fréquence et en type à ceux observés chez les patients adultes [voir AVERTISSEMENTS AND PRÉCAUTIONS Effets indésirables ]. Important findings et differences from adults are discussed in the following paragraphs.
Dans l'étude, Jia-i Humira a été étudié chez 171 patients qui avaient 4 à 17 ans avec JIA polyarticulaire. Les effets indésirables sévères rapportés dans l'étude comprenaient une pharyngite streptococcique de neutropénie, une augmentation de la métrorragie et de l'appendicite d'herpès zoster myosite métrorragie et d'appendicite. Des infections graves ont été observées chez 4% des patients dans les 2 ans suivant le début du traitement par Humira et comprenaient des cas de pharyngite infection des voies urinaires de pneumonie de l'herpès simplex et de zoster de l'herpès.
Dans l'étude, Jia-I 45% des patients ont connu une infection tout en recevant Humira avec ou sans MTX concomitante au cours des 16 premières semaines de traitement. Les types d'infections rapportés chez les patients traités par Humira étaient généralement similaires à ceux couramment observés chez les patients atteints de JIA polyarticulaire qui ne sont pas traités avec des bloqueurs de TNF. Lors de l'initiation du traitement, les réactions indésirables les plus courantes survenant dans cette population de patients traitées avec Humira ont été la douleur du site du site d'injection et la réaction du site d'injection (19% et 16% respectivement). Un événement indésirable moins communément signalé chez les patients recevant Humira était Granuloma Annulare qui n'a pas conduit à l'arrêt du traitement Humira.
Au cours des 48 premières semaines de traitement dans l'étude, des réactions d'hypersensibilité non sérieuses JIA-I ont été observées chez environ 6% des patients et comprenaient des réactions d'hypersensibilité allergique principalement localisées et une éruption allergique.
Dans l'étude, Jia-I 10% des patients traités par Humira qui avaient des anticorps anti-ADNdb négatifs ont développé des titres positifs après 48 semaines de traitement. Aucun patient n'a développé des signes cliniques d'auto-immunité pendant l'essai clinique.
Environ 15% des patients traités par Humira ont développé des élévations légères à modérées de la créatine phosphokinase (CPK) dans l'étude JIA-I. Des élévations dépassant 5 fois la limite supérieure de la normale ont été observées chez plusieurs patients. Les concentrations de CPK ont diminué ou retourné à la normale chez tous les patients. La plupart des patients ont pu continuer Humira sans interruption.
Dans l'étude, Jia-II Humira a été étudiée chez 32 patients qui étaient 2 à <4 years of age or 4 years of age et older weighing <15 kg with polyarticular JIA. The safety profile for this patient population was similar to the safety profile seen in patients 4 à 17 ans with polyarticular JIA.
Dans l'étude, JIA-II 78% des patients ont connu une infection lors de la réception de Humira. Ceux-ci comprenaient la nasopharyngite bronchite supérieure infection des voies respiratoires infectionnelles et étaient principalement légères à modérées en gravité. Des infections graves ont été observées chez 9% des patients recevant Humira dans l'étude et comprenaient une gastro-entérite et une varicelle rotavirus de caries dentaire.
Dans l'étude, des réactions allergiques non sérieuses JIA-II ont été observées chez 6% des patients et incluaient l'urticaire intermittente et les éruptions cutanées qui étaient toutes légères en gravité.
Arthrite psoriatique And Spondylarthrite ankylosante Études cliniques
has been studied in 395 patients with arthrite psoriatique (PsA) in two placebocontrolled trials et in an open lABel study et in 393 patients with spondylarthrite ankylosante (AS) in two placebo-controlled studies [voir Études cliniques ]. The safety profile for patients with PsA et AS treated with 40 mg toutes les deux semaines was similar to the safety profile seen in patients with RA Studies RA-I through IV.
Études cliniques de la maladie de Crohn
Adultes
Le profil de sécurité de Humira chez 1478 patients adultes atteints de la maladie de Crohn à partir de quatre études de prolongation contrôlées par placebo et de deux études d'extension ouverte [voir Études cliniques ] était similaire au profil de sécurité observé chez les patients atteints de PR.
Patients pédiatriques de 6 ans à 17 ans
Le profil de sécurité de Humira chez 192 patients pédiatriques d'une étude en double aveugle (étude PCD-I) et une étude d'extension ouverte [voir [voir Études cliniques ] était similaire au profil de sécurité observé chez les patients adultes atteints de la maladie de Crohn. Au cours de la phase d'induction de l'étiquette ouverte de 4 semaines, PCD-I, les réactions indésirables les plus courantes survenant dans la population pédiatrique traitée avec Humira étaient la douleur du site du site d'injection et la réaction du site d'injection (6% et 5% respectivement).
Au total, 67% des enfants ont connu une infection tout en recevant Humira dans l'étude PCD-I. Ceux-ci comprenaient une infection des voies respiratoires supérieures et une nasopharyngite.
Au total, 5% des enfants ont connu une infection grave tout en recevant Humira dans l'étude PCD-I. Ceux-ci comprenaient la septicémie liée à la septicémie (cathéter) gastro-entérite H1N1 et l'histoplasmose disséminée.
Dans l'étude, les réactions allergiques PCD-I ont été observées chez 5% des enfants qui étaient tous non sérives et étaient principalement des réactions localisées.
Rectocolite hémorragique Études cliniques
Adultes
Le profil de sécurité de Humira chez 1010 patients adultes atteints de colite ulcéreuse (UC) à partir de deux études contrôlées par placebo et une étude d'extension ouverte [voir Études cliniques ] était similaire au profil de sécurité observé chez les patients atteints de PR.
Patients pédiatriques de 5 ans à 17 ans
Le profil de sécurité de Humira chez 93 patients pédiatriques atteints de colite ulcéreuse d'une étude en double aveugle et une étude d'extension ouverte [voir Études cliniques ] était similaire au profil de sécurité observé chez les patients adultes atteints de colite ulcéreuse.
Psoriasis en plaques Études cliniques
has been studied in 1696 subjects with psoriasis en plaques (Ps) in placebo-controlled et open-lABel extension studies [voir Études cliniques ]. The safety profile for subjects with Ps treated with was similar to the safety profile seen in subjects with RA with the following exceptions. In the placebo-controlled portions of the clinical trials in Ps subjects -treated subjects had a higher incidence of arthralgia when comparouge to controls (3% vs. 1%).
Hidradénite suppurative Études cliniques
has been studied in 727 subjects with hidradenitis suppurativa (HS) in three placebocontrolled studies et one open-lABel extension study [voir Études cliniques ]. The safety profile for subjects with HS treated with weekly was consistent with the known safety profile of .
La fusée de HS définie comme une augmentation ≥25% par rapport à la référence dans les abcès et le nombre de nodules inflammatoires et avec un minimum de 2 lésions supplémentaires a été documentée dans 22 (22%) des 100 sujets qui ont été retirés du traitement Humira après le time-point d'efficacité primaire dans deux études.
Uvéite Études cliniques
has been studied in 464 adult patients with uveitis (UV) in placebo-controlled et open-lABel extension studies et in 90 pediatric patients with uveitis (Étudier PUV-i) [voir Études cliniques ]. Le profil de sécurité des patients atteints de UV traités avec Humira était similaire au profil de sécurité observé chez les patients atteints de PR.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, il existe un potentiel d'immunogénicité. La détection de la formation d'anticorps dépend fortement de la sensibilité et de la spécificité du test. De plus, l'incidence observée de l'anticorps (y compris les anticorps neutralisants) dans un test peut être influencée par plusieurs facteurs, notamment la méthodologie du test, le moment de la gestion des échantillons de médicaments concomitants de collecte d'échantillons et la maladie sous-jacente. Pour ces raisons, la comparaison de l'incidence des anticorps dans les études décrites ci-dessous avec l'incidence des anticorps dans d'autres études ou à d'autres produits d'adalimumab peut être trompeur.
Il existe deux tests qui ont été utilisés pour mesurer les anticorps anti-adalimumab. Avec les anticorps ELISA contre l'adalimumab ne pouvaient être détectés que lorsque les concentrations sériques d'adalimumab étaient <2 mcg/mL. The ECL assay can detect anti-adalimumab antibody titers independent of adalimumab concentrations in the serum samples. The incidence of antiadalimumab antibody (AAA) development in patients treated with are presented in TABle 2.
Tableau 2: Développement des anticorps anti-adalimumab déterminé par ELISA et test ECL chez les patients traités par Humira
| Indications | Durée de l'étude | Incidence des anticorps anti-adalimumab par ELISA (N / N) | Incidence des anticorps anti-adalimumab par test ECL (N / N) | ||
| Chez tous les patients qui ont reçu de l'adalimumab | Chez les patients atteints de concentrations sériques d'adalimumab <2 mcg/mL | ||||
| Polyarthrite rhumatoïde a | 6 à 12 mois | 5% (58/1062) | Non. | N / A | |
| Arthrite idiopathique juvénile (JIA) | 4 à 17 ans b | 48 semaines | 16% (27/171) | Non. | N / A |
| 2 à 4 ans ou ≥ 4 ans et peser <15 kg | 24 semaines | 7% (1/15) c | Non. | N / A | |
| Arthrite psoriatique d | 48 semaines e | 13% (24/178) | Non. | N / A | |
| Spondylarthrite ankylosante | 24 semaines | 9% (16/185) | Non. | N / A | |
| Maladie de Crohn adulte | 56 semaines | 3% (7/269) | 8% (7/86) | N / A | |
| La maladie de Crohn pédiatrique | 52 semaines | 3% (6/182) | 10% (6/58) | N / A | |
| Colite ulcéreuse adulte | 52 semaines | 5% (19/360) | 21% (19/92) | N / A | |
| Colite ulcéreuse pédiatrique | 52 semaines | 3% (3/100) | 13% (3/23) | 33% (33/100) i | |
| Psoriasis en plaques f | Jusqu'à 52 semaines g | 8% (77/920) | 21% (77/372) | N / A | |
| Hidradénite suppurative | 36 semaines | 7% (30/461) | 28% (58/207) h | 61% (272/445) j | |
| Uvéite non infectieuse | 52 semaines | 5% (12/249) | 21% (12/57) | 40% (99/249) k | |
| N: Nombre de patients atteints d'anticorps anti-adalimumab; NR: non signalé; NA: Non applicable (non effectué) a Chez les patients recevant du méthotrexate concomitant (MTX), l'incidence de l'anticorps anti-adalimumab était de 1% contre 12% avec la monothérapie Humira b Chez les patients recevant du MTX concomitant, l'incidence de l'anticorps anti-adalimumab était de 6% contre 26% avec la monothérapie Humira c Ce patient a reçu un MTX concomitant d Chez les patients recevant du MTX concomitant, l'incidence du développement des anticorps était de 7% contre 1% dans la PR e Sujets inscrits après avoir terminé 2 études précédentes de 24 semaines ou 12 semaines de traitements. f Chez le psoriasis en plaque, les patients atteints de monothérapie Humira et qui se retirent par la suite du traitement, le taux d'anticorps contre l'adalimumab après le retraitement était similaire au taux observé avant le retrait g Une étude de 12 semaines de phase 2 et une étude de 52 semaines de phase 3 h Parmi les sujets des 2 études de phase 3 qui ont arrêté le traitement Humira pendant une période pouvant aller jusqu'à 24 semaines et en qui les taux sériques de l'adalimumab ont ensuite refusé de <2 mcg/mL (approximately 22% of total subjects studied) i Aucune association apparente entre le développement et la sécurité des anticorps n'a été observée. L'association du développement des anticorps et de l'efficacité n'a pas été évaluée en raison du nombre limité de sujets dans chaque groupe de traitement stratifié par un titre d'anticorps anti-adalimumab. j Aucune association apparente entre le développement et la sécurité des anticorps n'a été observée k Aucune corrélation du développement des anticorps avec les résultats de sécurité ou d'efficacité n'a été observée |
Polyarthrite rhumatoïde And Arthrite psoriatique
Les patients en études RA-I RA-II et RA-III ont été testés à plusieurs moments pour les anticorps contre l'adalimumab en utilisant l'ELISA pendant la période de 6 à 12 mois. Aucune corrélation apparente du développement des anticorps aux effets indésirables n'a été observée. Avec des patients en monothérapie recevant toutes les deux semaines, le dosage peut développer des anticorps plus fréquemment que ceux qui reçoivent un dosage hebdomadaire. Chez les patients recevant la dose recommandée de 40 mg toutes les deux semaines comme monothérapie, la réponse de l'ACR 20 était plus faible chez les patients positifs aux anticorps que chez les patients néfastes des anticorps. L'immunogénicité à long terme de Humira est inconnue.
Expérience de commercialisation de la poste
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de Humira. Parce que ces réactions sont rapportées volontairement d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale à l'exposition à Humira.
Gastro-intestinal disorders: Diviculite de grandes perforations intestinales, y compris les perforations associées à la diverticulite et aux perforations appendiques associées à l'appendicite pancréatite
Troubles généraux et conditions du site d'administration: Pyrexie
Troubles hépato-biliaires: Hépatite d'insuffisance hépatique
Troubles du système immunitaire: Sarcoïdose
Néoplasmes bénins malins et non spécifiés (y compris les kystes et les polypes): Carcinome à cellules Merkel (carcinome neuroendocrinien de la peau)
Troubles du système nerveux: Troubles démyélinisants (par exemple la névrite optique Syndrome de Guillain-Barré) Accident cérébrovasculaire
Respiratoire disorders: Maladie pulmonaire interstitielle, y compris l'embolie pulmonaire de la fibrose pulmonaire
Réactions cutanées: Syndrome de Stevens Johnson Vascularite cutanée érythème multiforme ou aggravation du psoriasis (tous les sous-types, y compris la réaction cutanée pustuleuse et Palmoplantar)
Troubles vasculaires: Vascularite systémique thrombose veine profonde
Interactions médicamenteuses for Humira
Méthotrexate
has been studied in polyarthrite rhumatoïde (RA) patients taking concomitant methotrexate (MTX). Although MTX rougeuced the apparent adalimumab clearance the data ne pas suggest the need for dose adjustment of either or MTX [voir Pharmacologie clinique ].
Produits biologiques
Dans les études cliniques chez les patients atteints de PR, un risque accru d'infections graves a été observée avec la combinaison de bloqueurs de TNF avec Anakinra ou Abatacept sans avantage supplémentaire; Par conséquent, l'utilisation de Humira avec Abatacept ou Anakinra n'est pas recommandée chez les patients atteints de PR [voir AVERTISSEMENTS AND PRÉCAUTIONS ]. A higher rate of serious infections has also been observed in patients with RA treated with rituximAB who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of et other biologic products for the treatment of RA PsA AS CD UC Ps HS et UV. Concomitant administration of with other biologic DMARDS (e.g. anakinra et ABatacept) or other TNF blockers is pas recommended based upon the possible increased risk for infections et other potential pharmacological interactions.
Vaccins en direct
Évitez l'utilisation de vaccins vivants avec Humira [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Substrats du cytochrome P450
La formation d'enzymes CYP450 peut être supprimée par des concentrations accrues de cytokines (par exemple TNFα IL-6) pendant l'inflammation chronique. Il est possible pour une molécule qui antagonise l'activité des cytokines telles que l'adalimumab pour influencer la formation d'enzymes CYP450. Lors de l'initiation ou de l'arrêt de Humira chez les patients traités avec des substrats CYP450 avec une surveillance étroite de l'indice thérapeutique de l'effet (par exemple la warfarine) ou une concentration de médicament (par exemple, la cyclosporine ou la théophylline) est recommandée et la dose individuelle du médicament peut être ajustée au besoin.
Avertissements pour Humira
Inclus dans le cadre du PRÉCAUTIONS section.
Précautions pour Humira
Infections graves
Les patients traités par Humira présentent un risque accru de développer des infections graves impliquant divers systèmes d'organes et sites qui peuvent entraîner une hospitalisation ou la mort. Des infections opportunistes dues à des mycobactéries bactériennes invasives virales fongiques parasites ou à d'autres agents pathogènes opportunistes, notamment l'aspergillose blastomycosis candidose coccidiodomycose histoplasmosis la légionellose listériose pneumocystose et tuberculose ont été rapportées avec des bloqueurs TNF. Les patients ont fréquemment présenté une maladie disséminée plutôt que localisée.
L'utilisation concomitante d'un bloqueur TNF et d'un abatacept ou d'anakinra a été associée à un risque plus élevé d'infections graves chez les patients atteints de polyarthrite rhumatoïde (RA); Par conséquent, l'utilisation concomitante de Humira et de ces produits biologiques n'est pas recommandée dans le traitement des patients atteints de PR [voir AVERTISSEMENTS AND PRÉCAUTIONS et Interactions médicamenteuses ].
Le traitement par Humira ne doit pas être initié chez les patients atteints d'une infection active comprenant des infections localisées. Les patients de 65 ans et les patients plus âgés atteints de conditions comorbides et / ou de patients prenant des immunosuppresseurs concomitants (tels que les corticostéroïdes ou le méthotrexate) peuvent être plus à risque d'infection. Considérez les risques et les avantages du traitement avant de lancer un traitement chez les patients:
- avec une infection chronique ou récurrente;
- qui ont été exposés à la tuberculose;
- avec une histoire d'une infection opportuniste;
- qui ont résidé ou voyagé dans des zones de tuberculose endémique ou de mycoses endémiques telles que l'histoplasmose coccidiomidomycose ou la blastomycose; ou
- avec des conditions sous-jacentes qui peuvent les prédisposer à l'infection.
Tuberculose
Des cas de réactivation de la tuberculose et de nouvelles infections à la tuberculose à début ont été signalés chez les patients recevant Humira, y compris les patients qui ont déjà reçu un traitement pour la tuberculose latente ou active. Les rapports comprenaient des cas de tuberculose pulmonaire et extrapulmonaire (c'est-à-dire disséminé). Évaluer les patients pour les facteurs de risque de tuberculose et tester une infection latente avant de lancer Humira et périodiquement pendant le traitement.
Il a été démontré que le traitement de l'infection à la tuberculose latente avant le traitement par des agents de blocage du TNF réduit le risque de réactivation de la tuberculose pendant le traitement. Avant de lancer Humira, évaluez si le traitement de la tuberculose latente est nécessaire; et considérer une induration ≥ 5 mm un résultat positif de test cutané de la tuberculine même pour les patients auparavant vaccinés avec Bacille Calmette- guerin (BCG).
Considérons le traitement anti-tuberculose avant l'initiation de Humira chez les patients ayant des antécédents de tuberculose latente ou active dans qui un traitement adéquat ne peut pas être confirmé et pour les patients avec un test négatif pour la tuberculose latente mais ayant des facteurs de risque d'infection à la tuberculose. Malgré un traitement prophylactique de la tuberculose, des cas de tuberculose réactivés se sont produits chez les patients traités par Humira. La consultation avec un médecin ayant une expertise dans le traitement de la tuberculose est recommandée pour aider à décider si le début du traitement antituberculose est approprié pour un patient individuel.
Considérez fortement la tuberculose dans le diagnostic différentiel chez les patients qui développent une nouvelle infection pendant le traitement Humira, en particulier chez les patients qui ont déjà ou récemment voyagé dans des pays avec une prévalence élevée de tuberculose ou qui ont eu un contact étroit avec une personne atteinte de tuberculose active.
Surveillance
Surveillez de près les patients pour le développement de signes et symptômes d'infection pendant et après le traitement par Humira, notamment le développement de la tuberculose chez les patients qui ont testé négatif pour une infection latente tuberculose avant le début du traitement. Les tests d'infection à la tuberculose latente peuvent également être faussement négatifs lors de la thérapie avec Humira.
Arrêtez Humira si un patient développe une infection grave ou une septicémie. For a patient who develops a new infection during treatment with closely monitor them perform a prompt et complete diagnostic workup appropriate for an immunocompromised patient et initiate appropriate antimicrobial therapy.
Infections fongiques invasives
Si les patients développent une maladie systémique grave et qu'ils résident ou voyagent dans des régions où les mycoses sont endémiques, envisagez une infection fongique invasive dans le diagnostic différentiel. Les tests d'antigène et d'anticorps pour l'histoplasmose peuvent être négatifs chez certains patients atteints d'une infection active. Considérons le traitement antifongique empirique approprié en tenant compte à la fois du risque d'infection fongique sévère et des risques de thérapie antifongique pendant qu'un bilan diagnostique est effectué. Pour aider à la gestion de ces patients, envisagez une consultation avec un médecin ayant une expertise dans le diagnostic et le traitement des infections fongiques invasives.
Tumeurs malignes
Considérez les risques et les avantages du traitement du bloqueur TNF, notamment Humira avant de lancer un traitement chez les patients atteints d'une tumeur maligne connue autre qu'un cancer de la peau non mélanome traité avec succès (NMSC) ou lorsqu'il envisage de poursuivre un bloqueur de TNF chez les patients qui développent une maligne.
Tumeurs malignes In Adultes
Dans les parties contrôlées des essais cliniques de certains blocs de TNF, y compris Humira, plus de cas de tumeurs malignes ont été observés chez les patients adultes traités au bloc TNF par rapport aux patients adultes traités au contrôle. Pendant les parties contrôlées de 39 essais cliniques mondiaux de Humira chez des patients adultes atteints de polyarthrite rhumatoïde (RA) arthrite psoriasique (PSA) ankylosing (AS) Crohtis de Crohn (CD) Ulcéreuse (UC) Psoriasis (PS) Hiladenite Suppuratva (HS) et UVEITION (UV) que le malignes autre que la Psoritique (UV) que le MILIGNIGSE (HIGIGNIS Un cancer de la peau non mélanome (cellule basale et cellulaire épidermoïde) a été observé à un taux (intervalle de confiance à 95%) de 0,7 (NULL,48 1,03) pour 100 années de patient parmi 7973 patients traités par Humira contre 0,7 (NULL,41 1,17) pour 100 ans parmi les patients atteints de 4848 patients traités pour les patients atteints de 4 mois. Dans 52 essais cliniques contrôlés et incontrôlés globaux de Humira chez les patients adultes atteints de PSA RA comme CD UC PS HS et UV Les tumeurs malignes les plus fréquemment observées autres que le lymphome et le NMSC étaient le poumon et le mélanome de la prostate du côlon mammaire. Les tumeurs malignes chez les patients traités par Humira dans les parties contrôlées et incontrôlées des études étaient similaires de type et de nombre à ce qui serait attendu dans la population générale américaine selon la base de données SEER (ajustée pour le sexe et la race pour l'âge) .1 .1.
Dans les essais contrôlés d'autres bloqueurs du TNF chez les patients adultes à risque plus élevé de tumeurs malignes (c'est-à-dire les patients atteints de MPOC avec des antécédents de tabagisme significatifs et des patients traités au cyclophosphamide atteints de granulomatose de Wegener), une plus grande partie des tumeurs malignes s'est produite dans le groupe de bloqueurs TNF par rapport au groupe témoin.
Cancer de la peau non-mélanome
Pendant les parties contrôlées de 39 essais cliniques mondiaux Humira chez les patients adultes atteints de PSA RA comme CD UC PS HS et UV, le taux (intervalle de confiance à 95%) de NMSC était de 0,8 (NULL,52 1,09) pour 100 années de patient parmi les patients traités par Humira et 0,2 (NULL,10 0,59) pour 100 patients-années chez les patients traités par contrôle. Examinez tous les patients et en particulier les patients ayant des antécédents médicaux de traitement par immunosuppresseur prolongé antérieur ou les patients atteints de psoriasis ayant des antécédents de traitement PUVA pour la présence de NMSC avant et pendant le traitement par Humira.
Lymphome et leucémie
Dans les parties contrôlées des essais cliniques de tous les bloqueurs TNF chez les adultes, plus de cas de lymphome ont été observés chez les patients traités au bloc TNF par rapport aux patients traités avec le contrôle. Dans les parties contrôlées de 39 essais cliniques globaux de Humira chez des patients adultes atteints de PSA RA comme CD UC PS HS et les lymphomes UV 2 se sont produits chez 7973 patients traités par Humira contre 1 parmi 4848 patients traités au contrôle. Dans 52 essais cliniques contrôlés et incontrôlés globaux de Humira chez les patients adultes atteints de PSA RA comme CD UC PS HS et UV avec une durée médiane d'environ 0,7 ans, dont 24605 patients et plus de 40215 patients de Humira, le taux observé de lymphomes était d'environ 0,11 pour 100 patients. Ceci est environ 3 fois plus élevé que prévu dans la population générale américaine selon la base de données SEER (ajustée pour le sexe et la race d'âge) .1 Taux de lymphome dans les essais cliniques de Humira ne peuvent pas être comparés aux taux de lymphome dans les essais cliniques d'autres bloqueurs TNF et peuvent ne pas prédire les taux observés dans une population de patients plus large. Les patients atteints de PR et d'autres maladies inflammatoires chroniques, en particulier celles atteints de maladies très actives et / ou d'exposition chronique, aux thérapies par immunosuppresseurs peuvent être plus à risque (jusqu'à plusieurs intérêts) que la population générale pour le développement du lymphome même en l'absence de bloqueurs TNF. Des cas post-marketing de leucémie aiguë et chronique ont été signalés en association avec l'utilisation du blocage TNF dans la PR et d'autres indications. Même en l'absence de thérapie de traitement du TNF, les patients atteints de PR peuvent être à un risque plus élevé (environ 2 fois) que la population générale pour le développement de la leucémie.
Tumeurs malignes In Patients pédiatriques And Young Adultes
Tumeurs malignes some fatal have been reported among children adolescents et young adults who received treatment with TNF-blockers (initiation of therapy ≤ 18 years of age) of which is a member. Approximately half the cases were lymphomas including Hodgkin's et non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies et included rare malignancies usually associated with immunosuppression et malignancies that are pas usually observed in children et adolescents. The malignancies occurrouge after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing et are derived from a variety of sources including registries et spontaneous postmarketing reports.
Cas post-commercialisation de lymphome à cellules T hépatospléniques (HSTCL) Un type rare de lymphome à cellules T a été signalé chez des patients traités par des bloqueurs TNF, dont Humira. Ces cas ont eu une évolution de la maladie très agressive et ont été mortels. La majorité des cas de bloqueurs TNF signalés se sont produits chez les patients atteints de la maladie de Crohn ou de colite ulcéreuse et la majorité était chez les adolescents et les jeunes adultes. Presque tous ces patients avaient reçu un traitement avec les immunosuppresseurs azathioprine ou la 6-mercaptopurine (6â €) en même temps qu'un bloqueur de TNF ou avant le diagnostic. Il n'est pas certain que l'occurrence de HSTCL soit liée à l'utilisation d'un bloqueur TNF ou d'un bloqueur TNF en combinaison avec ces autres immunosuppresseurs. Le risque potentiel avec la combinaison de l'azathioprine ou de la 6mercaptopurine et de Humira doit être soigneusement considéré.
Réactions d'hypersensibilité
L'anaphylaxie et l'œdème angioneurotique ont été signalés après l'administration Humira. Si une réaction allergique anaphylactique ou autre se produit, arrêtez immédiatement l'administration de Humira et instituent une thérapie appropriée. Dans les essais cliniques des réactions d'hypersensibilité Humira (par exemple, réaction anaphylactoïde, réaction de médicament fixe, réaction médicamenteuse non spécifiée urticaire) ont été observées.
Réactivation du virus de l'hépatite B
L'utilisation de bloqueurs TNF, y compris Humira, peut augmenter le risque de réactivation du virus de l'hépatite B (HBV) chez les patients qui sont des porteurs chroniques de ce virus. Dans certains cas, la réactivation du VHB se produisant en conjonction avec le traitement des bloqueurs TNF a été fatale. La majorité de ces rapports se sont produits chez les patients recevant de manière concomitante d'autres médicaments qui suppriment le système immunitaire qui peut également contribuer à la réactivation du VHB. Évaluer les patients à risque d'infection par le VHB pour des preuves antérieures d'une infection par le VHB avant de lancer un traitement des bloqueurs TNF. Exercer une prudence dans la prescription des bloqueurs du TNF pour les patients identifiés comme porteurs de VHB. Des données adéquates ne sont pas disponibles sur l'innocuité ou l'efficacité du traitement des patients qui sont porteurs de VHB avec un traitement antiviral en conjonction avec le traitement des bloqueurs TNF pour prévenir la réactivation du VHB. Pour les patients qui sont porteurs de VHB et ont besoin d'un traitement avec des bloqueurs TNF, surveillez étroitement ces patients pour les signes cliniques et en laboratoire d'une infection active du VHB tout au long du traitement et pendant plusieurs mois suivant la résiliation du traitement. Chez les patients qui développent la réactivation du VHB arrêtent Humira et lancent un traitement antiviral efficace avec un traitement de soutien approprié. La sécurité de la reprise du traitement des bloqueurs TNF après le contrôle de la réactivation du VHB n'est pas connue. Par conséquent, faire preuve de prudence lorsque vous envisagez de reprise de la thérapie Humira dans cette situation et surveillez de près les patients.
Réactions neurologiques
L'utilisation d'agents de blocage du TNF, y compris Humira, a été associée à de rares cas de nouvel apparition ou d'exacerbation de symptômes cliniques et / ou de preuves radiographiques de la maladie démyélinisante du système nerveux central, notamment la sclérose en plaques (SEP) et la névrite optique et la maladie démyélinante périphérique, notamment le syndrome de Guillain-Barr. Exercer une prudence en considérant l'utilisation de Humira chez les patients atteints de troubles démyélinisants du système nerveux central ou périphérique à apparition récente ou à apparition récente; L'arrêt de Humira doit être pris en considération si l'un de ces troubles se développe. Il existe une association connue entre l'uvéite intermédiaire et les troubles démyélinisants centraux.
Réactions hématologiques
Des rapports rares de pancytopénie, y compris une anémie aplasique, ont été signalés avec des agents de blocage du TNF. Les effets indésirables du système hématologique, y compris la cytopénie médicalement significative (par exemple, la thrombocytopénie leukopénie) ont été rarement rapportés avec Humira. La relation causale de ces rapports à Humira n'est pas claire. Conseillez tous les patients de consulter des soins médicaux immédiats s'ils développent des signes et des symptômes suggérant des dyscrasies sanguines ou une infection (par exemple, une palette de saignement de fièvre persistante) lors de l'humira. Envisagez l'arrêt de la thérapie Humira chez les patients présentant des anomalies hématologiques significatives.
Risque accru d'infection lorsqu'il est utilisé avec Anakinra
L'utilisation concomitante d'Anakinra (un antagoniste de l'interleukine-1) et un autre bloqueur TNF a été associée à une plus grande proportion d'infections graves et de neutropénie et aucun avantage supplémentaire par rapport au bloqueur TNF seul chez les patients atteints de PR. Par conséquent, la combinaison de Humira et Anakinra n'est pas recommandée [voir Interactions médicamenteuses ].
Insuffisance cardiaque
Des cas d'aggravation d'insuffisance cardiaque congestive (CHF) et de nouveaux CHF à début ont été signalés avec des bloqueurs TNF. Des cas d'aggravation de CHF ont également été observés avec Humira. Humira n'a pas été officiellement étudié chez les patients atteints de CHF; Cependant, dans les essais cliniques d'un autre bloqueur de TNF, un taux plus élevé d'effets indésirables graves liés au CHF a été observé. Faites preuve de prudence lorsque vous utilisez Humira chez les patients souffrant d'insuffisance cardiaque et les surveillez attentivement.
Auto-immunité
Le traitement avec Humira peut entraîner la formation d'auto-anticorps et rarement dans le développement d'un syndrome de type lupus. Si un patient élabore des symptômes suggérant un syndrome de type lupus après un traitement par Humira arrête le traitement [voir Effets indésirables ].
Vaccination
Dans un essai clinique contrôlé par placebo des patients atteints de PR, aucune différence n'a été détectée dans la réponse des anticorps antipneumococciques entre les groupes de traitement Humira et le placebo lorsque le vaccin polysaccharide pneumococcal et le vaccin influencé a été administré parallèlement à Humira. Des proportions similaires de patients ont développé des niveaux protecteurs d'anticorps anti-influenza entre les groupes de traitement Humira et placebo; Cependant, les titres des antigènes agrégés aux grippe étaient modérément plus faibles chez les patients recevant Humira. La signification clinique de ceci est inconnue. Les patients sous Humira peuvent recevoir des vaccinations simultanées à l'exception des vaccins vivants. Aucune donnée n'est disponible sur la transmission secondaire de l'infection par les vaccins vivants chez les patients recevant Humira.
Il est recommandé que les patients pédiatriques soient si possibles à jour avec toutes les vaccinations en accord avec les directives actuelles de la vaccination avant de lancer une thérapie Humira. Les patients sous Humira peuvent recevoir des vaccinations simultanées à l'exception des vaccins vivants.
La sécurité de l'administration des vaccins vivant ou atténués chez les nourrissons exposés à Humira utero est inconnu. Les risques et les avantages doivent être pris en compte avant de vaccination (vivant ou liveté) pour nourrissons exposés [voir Utiliser dans des populations spécifiques ].
Risque accru d'infection lorsqu'il est utilisé avec l'abatacept
Dans les essais contrôlés, l'administration simultanée de bloqueurs TNF et d'abatacept a été associée à une plus grande proportion d'infections graves que l'utilisation d'un bloqueur TNF seul; La thérapie combinée par rapport à l'utilisation d'un bloqueur TNF à elle seule n'a pas démontré une amélioration des avantages cliniques dans le traitement de la PR. Par conséquent, la combinaison de l'abatacept avec des bloqueurs TNF, y compris Humira, n'est pas recommandée [voir Interactions médicamenteuses ].
Informations de conseil des patients
Conseillez au patient ou au soignant de lire l'étiquetage des patients approuvé par la FDA ( Guide des médicaments et Instructions pour une utilisation ).
Infections
Informer les patients que Humira peut réduire la capacité de leur système immunitaire à lutter contre les infections. Instruisez les patients de l'importance de contacter leur médecin s'ils développent des symptômes d'infection, y compris les infections fongiques invasives de la tuberculose et la réactivation des infections du virus de l'hépatite B [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Tumeurs malignes
Conseiller les patients du risque de tumeurs malignes lors de la réception de Humira [voir AVERTISSEMENTS AND PRÉCAUTIONS ]
Réactions d'hypersensibilité
Conseiller aux patients de consulter des soins médicaux immédiats s'ils éprouvent des symptômes de réactions d'hypersensibilité sévères. Conseiller les patients sensibles au latex que le capuchon d'aiguille de l'humira 40 mg / 0,8 ml de stylo et 40 mg / 0,8 ml 20 mg / 0,4 ml et 10 mg / 0,2 ml de la seringue préfabillée peut contenir le latex en caoutchouc naturel [voir AVERTISSEMENTS AND PRÉCAUTIONS Comment fourni / Stockage et manipulation ].
Autre Medical Conditions
Conseiller aux patients de signaler tout signe d'aggravation ou d'aggravation des conditions médicales telles que l'insuffisance cardiaque congestive, les troubles auto-immunes ou les cytopénies. Conseiller aux patients de signaler tout symptôme suggérant une cytopénie telle que des saignements meurtriers ou de la fièvre persistante [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Instructions sur la technique d'injection
Informez les patients que la première injection doit être effectuée sous la supervision d'un professionnel de la santé qualifié. Si un patient ou un soignant doit administrer Humira, instruisez-le dans les techniques d'injection et évaluez sa capacité à injecter par voie sous-cutanée pour assurer la bonne administration de Humira [voir Instructions pour une utilisation ].
Pour les patients qui utiliseront le stylo Humira leur disent qu'ils:
- Entendra un «click» fort lorsque le bouton d'activateur de couleur prune est enfoncé. Le clic fort signifie le début de l'injection.
- Doit continuer à tenir le stylo Humira contre leur peau surélevée pressée jusqu'à ce que tous les médicaments soient injectés. Cela peut prendre jusqu'à 15 secondes.
- Saura que l'injection s'est terminée lorsque le marqueur jaune apparaît complètement dans la vue de la fenêtre et cesse de se déplacer.
Demandez aux patients de disposer de leurs aiguilles et de seringues utilisés ou du stylo utilisé dans un conteneur d'élimination des objets tranchants transportés par la FDA immédiatement après l'utilisation. Demandez aux patients de ne pas éliminer les aiguilles et les seringues ou le stylo dans leurs ordures domestiques. Instruisez les patients que s'ils n'ont pas de conteneur d'élimination des objets tranchants approuvé par la FDA, ils peuvent utiliser un récipient domestique en plastique lourd peuvent être fermés avec un couvercle serré et résistant à la ponction sans les objets tranchants pour pouvoir distribuer des déchets dangereux à l'intérieur du contenant.
Instruisez les patients que lorsque leur conteneur d'élimination des objets tranchants est presque plein, ils devront suivre leurs directives communautaires pour la bonne façon de disposer de leur conteneur d'élimination des objets tranchants. Instruisez les patients qu'il peut y avoir des lois étatiques ou locales concernant l'élimination des aiguilles et des seringues utilisées. Reportez-vous aux patients sur le site Web de la FDA à https://www.fda.gov/safesharpsdisposal pour plus d'informations sur la disposition des Sharps sûrs et pour des informations spécifiques sur l'élimination des objets de barres dans l'État dans lesquelles ils vivent.
Demandez aux patients de ne pas disposer de leur conteneur d'élimination des objets tranchants utilisés dans leurs ordures ménagères, sauf si leurs directives communautaires le permettent. Demandez aux patients de ne pas recycler leur conteneur d'élimination des objets tranchants utilisés.
Toxicologie non clinique
Carcinogenèse Mutagenèse A trouble de la fertilité
Les études animales à long terme de Humira n'ont pas été menées pour évaluer le potentiel cancérigène ou son effet sur la fertilité.
Utiliser dans des populations spécifiques
Grossesse
Résumé des risques
Les études disponibles avec l'utilisation de l'adalimumab pendant la grossesse n'établissent pas de manière fiable une association entre l'adalimumab et les principales malformations congénitales. Les données cliniques sont disponibles auprès de l'Organisation des spécialistes de l'information sur la tératologie (OTIS) / Registre de grossesse MotherTobaby Humira chez les femmes enceintes atteintes de polyarthrite rhumatoïde (RA) ou de la maladie de Crohn (CD). Les résultats du registre ont montré un taux de 10% pour les principaux malformations congénitales avec une utilisation du premier trimestre de l'adalimumab chez les femmes enceintes atteintes de PR ou de CD et un taux de 7,5% pour les principales malformations congénitales dans la cohorte de comparaison catalysée. L'absence de schéma de malformations congénitales majeures est rassurante et les différences entre les groupes d'exposition peuvent avoir eu un impact sur la survenue de malformations congénitales (voir Données ).
L'adalimumab est activement transféré à travers le placenta au cours du troisième trimestre de la grossesse et peut affecter la réponse immunitaire chez le nourrisson exposé in-utera (voir Considérations cliniques ). In an embryo-fetal perinatal development study conducted in cynomolgus monkeys no fetal harm or malformations were observed with intravenous administration of adalimumab during organogenesis et later in gestation at doses that produced exposures up to approximately 373 times the maximum recommended human dose (MRHD) of 40 mg subcutaneous without methotrexate (see Données ).
Le risque de fond estimé des principaux malformations congénitales et une fausse couche pour les populations indiquées est inconnue. Toutes les grossesses présentent un risque de fond de perte de maltraitance natale ou d'autres résultats indésirables. Dans la population générale américaine, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est respectivement de 2 à 4% et 15 à 20%.
Considérations cliniques
Risque maternel et embryonnaire / fœtal associé à la maladie
Les données publiées suggèrent que le risque de résultats indésirables de la grossesse chez les femmes atteintes d'une PR ou d'une maladie inflammatoire de l'intestin (MII) est associée à une augmentation de l'activité de la maladie. Les résultats défavorables de la grossesse comprennent l'accouchement prématuré (avant 37 semaines de gestation) un faible poids à la naissance (moins de 2500 g) et un petit âge gestationnel à la naissance.
Réactions indésirables fœtales / néonatales
Les anticorps monoclonaux sont de plus en plus transportés à travers le placenta à mesure que la grossesse progresse avec la plus grande quantité transférée au cours du troisième trimestre (voir Données ). Risks et benefits should be considerouge prior to administering live or live-attenuated vaccines to infants exposed to utero [voir Utiliser dans des populations spécifiques ].
Données
Données humaines
Un registre exposition à la grossesse de cohorte prospective mené par Otis / MotherTobaby aux États-Unis et au Canada entre 2004 et 2016 a comparé le risque de malformations congénitales majeures chez les nourrissons nés vivants de 221 femmes (69 RA 152 CD) traitées avec de l'adalimumab au cours du premier trimestre et 106 femmes (74 RA 32 CD) non traitées par l'adalimumab.
La proportion de malformations congénitales majeures chez les nourrissons nés vivants dans les cohortes traitées à l'adalimumab et non traitées était de 10% (NULL,7% de CD de 10,5%) et 7,5% (NULL,8% RA 9,4% CD) respectivement. L'absence de schéma de malformations congénitales majeures est rassurante et les différences entre les groupes d'exposition peuvent avoir eu un impact sur la survenue de malformations congénitales. Cette étude ne peut pas déterminer de manière fiable s'il existe une association entre l'adalimumab et les principales malformations congénitales en raison des limites méthodologiques du registre, y compris une petite taille de l'échantillon, la nature volontaire de l'étude et la conception non randomisée.
Dans une étude clinique indépendante menée chez dix femmes enceintes atteintes de MII traitées avec des concentrations d'adalimumab Humira a été mesurée dans le sérum maternel ainsi que dans le sang de cordon (n = 10) et le sérum infantile (n = 8) le jour de la naissance. La dernière dose de Humira a été donnée entre 1 et 56 jours avant la livraison. Les concentrations d'adalimumab étaient de 0,16-19,7 μg / ml dans du sang de cordon 4,28-17,7 μg / ml dans le sérum infantile et 0-16,1 μg / ml dans le sérum maternel. Dans tous les cas sauf un, la concentration sanguine du cordon de l'adalimumab était plus élevée que la concentration sérique maternelle suggérant que l'adalimumab traverse activement le placenta. De plus, un nourrisson avait des concentrations sériques à chacune des éléments suivants: 6 semaines (NULL,94 μg / ml) 7 semaines (NULL,31 μg / ml) 8 semaines (NULL,93 μg / ml) et 11 semaines (NULL,53 μg / ml) suggérant que l'adalimumab peut être détecté dans le sérum de nourrissons exposés in utero pendant au moins 3 mois de la naissance.
Données sur les animaux
Dans une étude de développement périnatal embryo-fœtal, les singes de Cynomolgus enceintes ont reçu l'adalimumab des jours de gestation 20 à 97 à des doses qui produisaient des expositions jusqu'à 373 fois celles qui ont atteint le MRHD sans méthotrexate (sur une base AUC avec des doses IV maternelles jusqu'à 100 mg / kg / semaine). L'adalimumab n'a pas causé de dommage aux fœtus ou aux malformations.
Lactation
Résumé des risques
Des données limitées des rapports de cas dans la littérature publiée décrivent la présence de l'adalimumab dans le lait maternel à des doses infantiles de 0,1% à 1% de la concentration sérique maternelle. Les données publiées suggèrent que l'exposition systémique à un nourrisson allaité devrait être faible car l'adalimumab est une grande molécule et est dégradé dans le tractus gastro-intestinal. Cependant, les effets de l'exposition locale dans le tractus gastro-intestinal sont inconnus. Il n'y a aucun rapport d'effets néfastes de l'adalimumab sur le nourrisson allaité et aucun effet sur la production de lait. Les avantages du développement et de la santé de l'allaitement doivent être pris en compte avec le besoin clinique de la mère pour Humira et tout effet indésirable potentiel sur l'enfant allaité de Humira ou de l'état maternel sous-jacent.
Usage pédiatrique
La sécurité et l'efficacité de Humira ont été établies pour:
- Réduire les signes et symptômes de JIA polyarticulaire modérément à gravement active chez les patients pédiatriques de 2 ans et plus.
- Le traitement de la maladie de Crohn modérément à gravement active chez les patients pédiatriques de 6 ans et plus.
- Le traitement de la colite ulcéreuse modérément à sévèrement active chez les patients pédiatriques de 5 ans et plus.
- Le traitement de la Hidradénite suppurativa modérée à sévère chez les patients âgés de 12 ans et plus.
- Le traitement de l'intermédiaire non infectieux et de la panuvéite chez les patients pédiatriques de 2 ans et plus.
En raison de son inhibition du TNFα humira administré pendant la grossesse pourrait affecter la réponse immunitaire chez le nouveau-né et le nourrisson exposé à utéro. Les données de huit nourrissons exposés à Humira in utero suggèrent que l'adalimumab traverse le placenta [voir Utiliser dans des populations spécifiques ]. The clinical significance of elevated adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks et benefits should be considerouge prior to vaccinating (live or live-attenuated) exposed infants.
Les cas de lymphome post-marketing, y compris le lymphome hépatosplénique à cellules T et d'autres tumeurs malignes, certains ont été signalés parmi les enfants adolescents et les jeunes adultes qui ont reçu un traitement avec des blocs de TNF, dont Humira [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Arthrite idiopathique juvénile
Dans l'étude, Jia-i Humira a réduit les signes et symptômes de la JIA polyarticulaire active chez les patients de 4 à 17 ans [voir Études cliniques ]. In Étudier Jia-iI the safety profile for patients 2 to <4 years of age was similar to the safety profile for patients 4 à 17 ans with polyarticular JIA [voir Effets indésirables ]. has pas been studied in patients with polyarticular JIA less than 2 years of age or in patients with a weight below 10 kg.
La sécurité de Humira chez les patients dans les essais polyarticulaires de JIA était généralement similaire à celle observée chez les adultes à certaines exceptions [voir Effets indésirables ].
La sécurité et l'efficacité de Humira n'ont pas été établies chez les patients pédiatriques atteints de JIA de moins de 2 ans.
Maladie pédiatrique de Crohn
La sécurité et l'efficacité de Humira pour le traitement de la maladie de Crohn modérément à gravement active ont été établies chez des patients pédiatriques de 6 ans et plus. L'utilisation de Humira pour cette indication est étayée par des preuves d'études adéquates et bien contrôlées chez des adultes avec des données supplémentaires provenant d'une étude clinique en double aveugle randomisée de 52 semaines de deux concentrations de dose de Humira chez 192 patients pédiatriques (6 ans à 17 ans) [voir Effets indésirables Pharmacologie clinique Études cliniques ]. The adverse reaction profile in patients 6 years to 17 years of age was similar to adults.
La sécurité et l'efficacité de Humira n'ont pas été établies chez les patients pédiatriques atteints de la maladie de Crohn de moins de 6 ans.
Colite ulcéreuse pédiatrique
La sécurité et l'efficacité de Humira pour le traitement d'une colite ulcéreuse modérément à sévère ont été établies chez des patients pédiatriques de 5 ans et plus. L'utilisation de Humira pour cette indication est étayée par des preuves d'études adéquates et bien contrôlées chez des adultes présentant des données supplémentaires provenant d'une étude clinique en double aveugle randomisée de 52 semaines de deux concentrations de dose de Humira chez 93 patients pédiatriques (5 à 17 ans) [voir Effets indésirables Pharmacologie clinique Études cliniques ]. The adverse reaction profile in patients 5 years to 17 years of age was similar to adults.
L'efficacité de Humira n'a pas été établie chez les patients qui ont perdu une réponse ou qui ont été intolérants aux bloqueurs du TNF.
La sécurité et l'efficacité de Humira n'ont pas été établies chez les patients pédiatriques atteints de colite ulcéreuse de moins de 5 ans.
Uvéite pédiatrique
La sécurité et l'efficacité de Humira pour le traitement de l'uvéite non infectieuse ont été établies chez les patients pédiatriques de 2 ans et plus. L'utilisation de Humira est étayée par des preuves d'études adéquates et bien contrôlées de Humira chez les adultes et une étude clinique contrôlée randomisée 2: 1 chez 90 patients pédiatriques [voir Études cliniques ]. The safety et effectiveness of have pas been estABlished in pediatric patients with uveitis less than 2 years of age.
Hidradénite suppurative
L'utilisation de Humira chez les patients pédiatriques de 12 ans et plus pour le HS est étayée par des preuves d'études adéquates et bien contrôlées de Humira chez les patients HS adultes. La modélisation et la simulation pharmacocinétiques de la population supplémentaires prédisaient que le dosage basé sur le poids de Humira chez les patients pédiatriques de 12 ans et plus peut fournir une exposition généralement similaire aux patients adultes HS. Le cours du HS est suffisamment similaire chez les patients adultes et adolescents pour permettre l'extrapolation des données de l'adulte aux patients adolescents. La dose recommandée chez les patients pédiatriques de 12 ans ou plus est basée sur le poids corporel [voir Posologie et administration Pharmacologie clinique et Études cliniques ].
La sécurité et l'efficacité de Humira n'ont pas été établies chez les patients de moins de 12 ans avec HS.
Utilisation gériatrique
Au total, 519 patients atteints de PR 65 ans et plus, dont 107 patients âgés de 75 ans et plus ont reçu Humira dans les études cliniques RA-I via IV. Aucune différence globale d'efficacité n'a été observée entre ces patients et les patients plus jeunes. La fréquence de l'infection grave et de la tumeur maligne chez les patients traités par Humira de 65 ans et plus était plus élevé que pour ceux de moins de 65 ans. Considérez les avantages et les risques de Humira chez les patients de 65 ans et plus. Chez les patients traités avec Humira, surveillez de près le développement d'une infection ou d'une malignité [voir AVERTISSEMENTS AND PRÉCAUTIONS ].
Informations sur la surdose pour Humira
Des doses allant jusqu'à 10 mg / kg ont été administrées aux patients dans des essais cliniques sans signe de toxicités limitant la dose. En cas de surdosage, il est recommandé de surveiller le patient pour tout signe ou symptôme de réactions indésirables ou d'effets et un traitement symptomatique approprié immédiatement.
Contre-indications pour Humira
Aucun.
Pharmacologie clinique for Humira
Mécanisme d'action
L'adalimumab se lie spécifiquement au TNF-alpha et bloque son interaction avec les récepteurs TNF de surface cellulaire p55 et p75. L'adalimumab lyse en surface TNF exprimant les cellules in vitro en présence de complément. L'adalimumab ne se lie ni n'inactive la lymphotoxine (TNF-beta). Le TNF est une cytokine naturelle qui est impliquée dans les réponses inflammatoires et immunitaires normales. Des concentrations élevées de TNF se trouvent dans le liquide synovial des patients atteints de RA Jia PSA et jouent un rôle important dans l'inflammation pathologique et la destruction articulaire qui sont les caractéristiques de ces maladies. Des concentrations accrues de TNF se trouvent également dans les plaques de psoriasis. Dans le traitement PS avec Humira peut réduire l'épaisseur épidermique et l'infiltration des cellules inflammatoires. La relation entre ces activités pharmacodynamiques et les mécanismes par lesquelles Humira exerce ses effets cliniques est inconnue.
L'adalimumab module également les réponses biologiques induites ou régulées par le TNF, y compris les changements dans les concentrations des molécules d'adhésion responsables de la migration des leucocytes (ELAM-1 VCAM-1 et ICAM-1 avec un IC50 de 1-2 x 10-10m).
Pharmacodynamique
Après le traitement avec Humira, une diminution des concentrations de réactifs de phase aiguë de l'inflammation (protéine C-réactive [CRP] et le taux de sédimentation des érythrocytes [ESR]) et les cytokines sériques (IL-6) ont été observées par rapport à la ligne de base chez les patients atteints de polyarthrite rhumatoïde. Une diminution des concentrations de CRP a également été observée chez les patients atteints de la colite ulcéreuse de la maladie de Crohn et de Hidradénite suppurativa. Les concentrations sériques de métalloprotéinases matricielles (MMP-1 et MMP-3) qui produisent un remodelage tissulaire responsable de la destruction du cartilage ont également été diminuées après l'administration de Humira.
Pour les patients pédiatriques de 5 ans à 17 ans avec une colite ulcéreuse, la dose recommandée de Humira est basée sur des relations dose / exposition-efficacité modélisées et des données pharmacocinétiques. Il n'y a aucune différence d'efficacité cliniquement pertinente anticipée entre la dose plus élevée étudiée dans l'essai clinique (semaines à 52 dans l'étude PUC-I) [voir Études cliniques ] et la posologie recommandée [voir Posologie et administration ].
Pharmacocinétique
La pharmacocinétique de l'adalimumab était linéaire sur la plage de dose de 0,5 à 10 mg / kg après l'administration d'une seule dose intraveineuse (Humira n'est pas approuvé pour un usage intraveineux). Après 20 40 et 80 mg toutes les deux semaines et chaque semaine Administration sous-cutanée Adalimumab Les concentrations de creux sériques moyennes à l'état d'équilibre ont augmenté approximativement proportionnellement à la dose chez les patients atteints de PR. La demi-vie terminale moyenne était d'environ 2 semaines allant de 10 à 20 jours à tous les études. Les sujets sains et les patients atteints de PR présentaient une pharmacocinétique d'adalimumab similaire.
L'exposition à l'adalimumab chez les patients traitées avec 80 mg toutes les deux semaines est estimée à celle des patients traités avec 40 mg chaque semaine.
Absorption
La biodisponibilité absolue moyenne de l'adalimumab après une seule dose sous-cutanée de 40 mg était de 64%. Le temps moyen pour atteindre la concentration maximale était de 5,5 jours (131 ± 56 heures) et la concentration sérique maximale était de 4,7 ± 1,6 mcg / ml chez des sujets sains après une seule administration sous-cutanée de 40 mg de Humira.
Distribution
Le volume de distribution (VSS) variait de 4,7 à 6,0 L après l'administration intraveineuse de doses allant de 0,25 à 10 mg / kg chez les patients atteints de PR.
Élimination
La pharmacocinétique à dose unique de l'adalimumab chez les patients atteints de PR a été déterminée dans plusieurs études avec des doses intraveineuses allant de 0,25 à 10 mg / kg. La clairance systémique de l'adalimumab est d'environ 12 ml / h. Dans les études à long terme avec le dosage de plus de deux ans, il n'y avait aucune preuve de changements dans la clairance au fil du temps chez les patients atteints de PR.
Population de patients
Polyarthrite rhumatoïde And Spondylarthrite ankylosante
Chez les patients recevant 40 mg de Humira toutes les deux semaines, les concentrations de creux à l'état d'équilibre de l'adalimumab étaient environ 5 mcg / ml et 8 à 9 mcg / ml sans et avec un traitement concomitant MTX respectivement. Les concentrations d'adalimumab dans le liquide synovial de cinq patients atteints de polyarthrite rhumatoïde variaient de 31 à 96% de celles en sérum. La pharmacocinétique de l'adalimumab chez les patients atteints de SA similaires à ceux des patients atteints de PR.
Arthrite psoriatique
Chez les patients recevant 40 mg toutes les deux semaines, l'adalimumab moyen que les concentrations de creux à l'état d'équilibre étaient de 6 à 10 mcg / ml et 8,5 à 12 mcg / ml sans et avec un traitement concomitant MTX respectivement.
Psoriasis en plaques
L'adalimumab signifie que la concentration en abondance à l'état d'équilibre était d'environ 5 à 6 mcg / ml pendant le traitement de Humira 40 mg toutes les deux semaines.
Uvéite adulte
L'adalimumab signifie que la concentration régulière était d'environ 8 à 10 mcg / ml pendant le traitement de Humira 40 mg toutes les deux semaines.
Hidradénite adulte suppurative
Les concentrations de l'après l'adalimumab étaient d'environ 7 à 8 mcg / ml à la semaine 2 et la semaine 4 respectivement après avoir reçu 160 mg à la semaine 0 suivi de 80 mg à la semaine 2. Les concentrations moyennes de l'auge en régime permanent à la semaine 12 à la semaine 36 étaient d'environ 7 à 11 mcg / ml pendant Humira 40 mg par semaine par semaine.
Maladie adulte de Crohn
Les concentrations moyennes d'adalimumab moyens étaient d'environ 12 mcg / ml à la semaine 2 et la semaine 4 après avoir reçu 160 mg à la semaine 0 suivi de 80 mg à la semaine 2. Les concentrations moyennes de creux à l'état d'équilibre étaient de 7 mcg / ml à la semaine 24 et la semaine 56 pendant le traitement de Humira 40 mg toutes les autres semaines.
Colite ulcéreuse adulte
Les concentrations moyennes de l'après l'adalimumab étaient d'environ 12 mcg / ml à la semaine 2 et la semaine 4 après avoir reçu 160 mg à la semaine 0 suivi de 80 mg à la semaine 2. Les concentrations moyennes de l'auge en régime permanent étaient d'environ 8 mcg / ml et 15 mcg / ml à la semaine 52 après avoir reçu une dose de Humira 40 mg toutes les deux semaines et 40 mg toutes les semaines respectivement.
Effets des anticorps anti-drogue sur la pharmacocinétique
Polyarthrite rhumatoïde
Une tendance vers une clairance apparente plus élevée de l'adalimumab en présence d'anticorps anti-adalimumab a été identifiée.
Colite ulcéreuse pédiatrique
Les anticorps contre l'adalimumab par essai ECL ont été associés à des concentrations sériques d'adalimumab réduites chez des patients pédiatriques atteints de colite ulcéreuse modérément à sévère.
Hidradénite suppurative
Chez les sujets présentant des anticorps HS modérés à sévères vers l'adalimumab, ont été associés à des concentrations sériques d'adalimumab réduites. En général, l'étendue de la réduction des concentrations sériques d'adalimumab est plus élevée avec des titres croissants d'anticorps contre l'adalimumab.
Populations spécifiques
Patients gériatriques
Une clairance inférieure avec l'âge croissant a été observée chez les patients atteints de RA âgés de 40 à 75 ans.
Patients pédiatriques
Arthrite idiopathique juvénile
- 4 ans à 17 ans: les concentrations de creux moyens de l'adalimumab en régime permanent étaient de 6,8 mcg / ml et 10,9 mcg / ml chez les patients pesant <30 kg receiving 20 mg subcutaneously every other week as mopasherapy or with concomitant MTX respectively. The adalimumab mean steady-state trough concentrations were 6.6 mcg/mL et 8.1 mcg/mL in patients weighing ≥30 kg receiving 40 mg subcutaneously every other week as mopasherapy or with MTX concomitant treatment respectively.
- 2 ans pour <4 years of age or 4 years of age et older weighing <15 kg: The adalimumab mean steady-state trough adalimumab concentrations were 6.0 mcg/mL et 7.9 mcg/mL in patients receiving subcutaneously every other week as mopasherapy or with MTX concomitant treatment respectively.
Hidradénite pédiatrique suppurativa
Les concentrations d'adalimumab chez les adolescents atteints de HS recevant les schémas posologiques recommandés devraient être similaires à celles observées chez des sujets adultes atteints de HS basée sur la modélisation et la simulation pharmacocinétiques de la population.
La maladie de Crohn pédiatrique
Les concentrations moyennes de l'adalimumab moyenne étaient de 15,7 ± 6,5 mcg / ml à la semaine 4 après 160 mg à la semaine 0 et 80 mg à la semaine 2 et 10,5 ± 6,0 mcg / ml à la semaine 52 après 40 mg toutes les deux semaines chez les patients avec des patients pesant ≥ 40 kg. Les concentrations moyennes de l'adalimumab ± SD étaient de 10,6 ± 6,1 mcg / ml à la semaine 4 après l'administration de 80 mg à la semaine 0 et 40 mg à la semaine 2 et 6,9 ± 3,6 mcg / ml <40 kg.
Colite ulcéreuse pédiatrique
La concentration de creux à l'état d'équilibre de l'adalimumab était de 5,0 ± 3,3 mcg / ml à la semaine 52 après l'administration sous-cutanée de 0,6 mg / kg (maximum de 40 mg) toutes les deux semaines chez les patients atteints de CU pédiatrique de 5 ans à 17 ans. Chez les patients qui ont reçu 0,6 mg / kg (maximum de 40 mg) chaque semaine, la concentration moyenne de creux à l'état d'équilibre était de 15,7 ± 5,6 mcg / ml à la semaine 52 chez les patients atteints de CU pédiatrique de 5 ans à 17 ans.
Patients masculins et féminins
Aucune différence pharmacocinétique liée au genre n'a été observée après correction du poids corporel d'un patient. Des sujets sains et des patients atteints de polyarthrite rhumatoïde présentaient une pharmacocinétique adalimumab similaire.
Patients souffrant de troubles rénaux ou hépatiques
Aucune donnée pharmacocinétique n'est disponible chez les patients souffrant de troubles hépatiques ou rénaux.
Facteur rhumatoïde ou concentrations de CRP
Des augmentations mineures de la clairance apparente ont été prédites chez les patients atteints de PR recevant des doses inférieures à la dose recommandée et chez les patients atteints de PR avec un facteur rhumatoïde élevé ou des concentrations de CRP. Ces augmentations ne sont pas susceptibles d'être cliniquement importantes.
Études d'interaction médicamenteuse
Méthotrexate
Le MTX a réduit la clairance apparente de l'adalimumab après un dosage unique et multiple de 29% et 44% respectivement chez les patients atteints de PR [voir Interactions médicamenteuses ].
Études cliniques
Polyarthrite rhumatoïde
L'efficacité et l'innocuité de Humira ont été évaluées dans cinq études randomisées en double aveugle chez des patients ≥ 18 ans avec une polyarthrite rhumatoïde active diagnostiquée selon les critères de l'American College of Rhumatology (ACR). Les patients avaient au moins 6 articulations gonflées et 9 tendres. Humira a été administré par voie sous-cutanée en combinaison avec le méthotrexate (MTX) (NULL,5 à 25 mg étudie RA-I RA-III et RA-V) ou en monothérapie (études RA-II et RA-V) ou avec d'autres médicaments anti-Rheumatic modifiant la maladie (DMARD) (étude RA-IV).
L'étude RA-I a évalué 271 patients qui avaient échoué un traitement avec au moins un mais pas plus de quatre ARM et ont eu une réponse inadéquate au MTX. Des doses de 20 40 ou 80 mg de Humira ou de placebo ont été données toutes les deux semaines pendant 24 semaines.
L'étude RA-II a évalué 544 patients qui avaient échoué un traitement avec au moins un DMARD. Des doses de placebo 20 ou 40 mg de Humira ont été données en monothérapie toutes les deux semaines ou chaque semaine pendant 26 semaines.
L'étude RA-III a évalué 619 patients qui ont eu une réponse inadéquate au MTX. Les patients ont reçu un placebo 40 mg de Humira toutes les deux semaines avec des injections de placebo sur des semaines alternatives ou 20 mg d'Humira chaque semaine jusqu'à 52 semaines. L'étude RA-III avait un critère d'évaluation primaire supplémentaire à 52 semaines d'inhibition de la progression de la maladie (comme détecté par les résultats des rayons X). À la fin des 52 premières semaines, 457 patients inscrits à une phase d'extension ouverte dans laquelle 40 mg de Humira ont été administrés toutes les deux semaines jusqu'à 5 ans.
L'étude RA-IV a évalué la sécurité chez 636 patients qui étaient soit naïfs de DMARD ou ont été autorisés à rester sur leur thérapie rhumatologique préexistante à condition que le traitement soit stable pendant au moins 28 jours. Les patients ont été randomisés pour 40 mg de Humira ou de placebo toutes les deux semaines pendant 24 semaines.
L'étude RA-V a évalué 799 patients atteints de PR modérément à sévère active de moins de 3 ans de durée qui avait ≥ 18 ans et Mtx Naã¯ve. Les patients ont été randomisés pour recevoir MTX (optimisé à 20 mg / semaine par semaine 8) Humira 40 mg toutes les deux semaines ou la thérapie combinée Humira / MTX pendant 104 semaines. Les patients ont été évalués pour les signes et symptômes et pour la progression radiographique des lésions articulaires. La durée médiane de la maladie chez les patients inscrits à l'étude était de 5 mois. La dose médiane de MTX obtenue était de 20 mg.
Réponse clinique
Le pourcentage de patients traités par Humira atteignant l'ACR 20 50 et 70 réponses dans les études RAII et III est présenté dans le tableau 3.
Tableau 3: Réponses ACR dans les études RA-II et RA-III (pour cent des patients)
| Réponse | Étude de la monothérapie RA-II (26 semaines) | Étude combinaison de méthotrexate RA-III (24 et 52 semaines) | |||
| Placebo N = 110 | 40 mg toutes les deux semaines N = 113 | 40 mg weekly N = 103 | Placebo/ MTX N = 200 | / MTX 40 mg toutes les deux semaines N = 207 | |
| Acr20 | |||||
| Mois 6 | 19% | 46% * | 53% * | 30% | 63% * |
| Mois 12 | N / A | N / A | N / A | 24% | 59% * |
| Acr50 | |||||
| Mois 6 | 8% | 22% * | 35% * | 10% | 39% * |
| Mois 12 | N / A | N / A | N / A | 10% | 42% * |
| ACR70 | |||||
| Mois 6 | 2% | 12%* | 18% * | 3% | 21% * |
| Mois 12 | N / A | N / A | N / A | 5% | 23% * |
| * P <0.01 vs. placebo |
Les résultats de l'étude RA-I étaient similaires à l'étude RA-III; Les patients recevant Humira 40 mg toutes les deux semaines dans l'étude RA-I a également atteint ACR 20 50 et 70 taux de réponse de 65% 52% et 24% respectivement par rapport aux réponses placebo de 13% 7% et 3% respectivement à 6 mois (P <0.01).
Les résultats des composants des critères de réponse de l'ACR pour les études RA-II et RA-III sont présentés dans le tableau 4. Les taux de réponse ACR et l'amélioration de toutes les composants de la réponse ACR ont été maintenus à la semaine 104. Au cours des 2 années d'étude, RA-III 20% des patients Humira réceptant 40 mg toutes les autres semaines sur une période clinique majeure définie comme l'entretien d'une réponse ACR 70 au cours d'une période de 6 mois. Les réponses ACR ont été maintenues dans des proportions similaires de patients pendant jusqu'à 5 ans avec un traitement à Humira continu dans la partie ouverte de l'étude RA-III.
Tableau 4: Composantes de la réponse ACR dans les études RA-II et RA-III
| Paramètre (médian) | Étude RA-II | Étude RA-III | ||||||
| Placebo N = 110 | a N = 113 | Placebo/ Mtx N = 200 | a / Mtx N = 207 | |||||
| Base de base | WK 26 | Base de base | WK 26 | Base de base | WK 24 | Base de base | WK 24 | |
| Nombre de joints tendres (0-68) | 35 | 26 | 31 | 16 * | 26 | 15 | 24 | 8 * |
| Nombre d'articulations gonflées (0-66) | 19 | 16 | 18 | 10 * | 17 | 11 | 18 | 5 * |
| Évaluation globale des médecins b | 7.0 | 6.1 | 6.6 | 3.7 * | 6.3 | 3.5 | 6.5 | 2.0 * |
| Évaluation globale des patients b | 7.5 | 6.3 | 7.5 | 4.5 * | 5.4 | 3.9 | 5.2 | 2.0 * |
| Douleur b | 7.3 | 6.1 | 7.3 | 4.1 * | 6.0 | 3.8 | 5.8 | 2.1 * |
| Indice de handicap (HAQ) c | 2.0 | 1.9 | 1.9 | 1.5 * | 1.5 | 1.3 | 1.5 | 0,8 * |
| CRP (Mg / DL) | 3.9 | 4.3 | 4.6 | 1.8 * | 1.0 | 0.9 | 1.0 | 0,4 * |
| a 40 mg Humira administré toutes les deux semaines b Échelle visuelle analogique; 0 = meilleur 10 = pire c Indice de handicap du questionnaire sur l'évaluation de la santé; 0 = Meilleur 3 = Pire mesures La capacité du patient à effectuer les éléments suivants: Vobe / marié ARIRE EAT WAKE RECKAT GRIP HAUP HYGIENE ET RETENU Activité quotidienne * P <0.001 vs. placebo based on mean change from baseline |
L'évolution dans le temps de la réponse ACR 20 pour l'étude RA-III est illustrée à la figure 1.
Dans l'étude, RA-III 85% des patients atteints de réponses ACR 20 à la semaine 24 ont maintenu la réponse à 52 semaines. L'évolution temporelle de la réponse ACR 20 pour l'étude RA-I et l'étude RA-II étaient similaires.
Figure 1: Étude RA-III ACR 20 réponses sur 52 semaines
 |
Dans l'étude, RA-IV 53% des patients traités avec Humira 40 mg toutes les deux semaines plus la norme de soins ont eu une réponse ACR 20 à la semaine 24, contre 35% sur placebo plus la norme de soins (P <0.001). No unique adverse reactions related to the combination of (adalimumab) et other DMARDs were observed.
Dans l'étude, RA-V avec des patients MTX Naã¯ve avec un début de début RA, le traitement combiné avec Humira plus MTX a conduit à des pourcentages plus élevés de patients atteignant des réponses ACR que la monothérapie MTX ou la monothérapie Humira à la semaine 52 et les réponses ont été maintenues à la semaine 104 (voir tableau 5).
Tableau 5: Réponse de l'ACR dans l'étude RA-V (pour cent des patients)
| Réponse | MTX b N = 257 | c N = 274 | / Mtx N = 268 |
| Acr20 | |||
| Semaine 52 | 63% | 54% | 73% |
| Semaine 104 | 56% | 49% | 69% |
| Acr50 | |||
| Semaine 52 | 46% | 41% | 62% |
| Semaine 104 | 43% | 37% | 59% |
| ACR70 | |||
| Semaine 52 | 27% | 26% | 46% |
| Semaine 104 | 28% | 28% | 47% |
| Réponse clinique majeure a | 28% | 25% | 49% |
| a La réponse clinique majeure est définie comme réalisant une réponse ACR70 pour une période continue de six mois b p <0.05 / Mtx vs. MTX for ACR 20 p <0.001 / Mtx vs. MTX for ACR 50 et 70 et Réponse clinique majeure c p <0.001 / Mtx vs. |
À la semaine 52, toutes les composantes individuelles des critères de réponse ACR pour l'étude RA-V se sont améliorées dans le groupe Humira / MTX et des améliorations ont été maintenues à la semaine 104.
Réponse radiographique
Dans l'étude, les lésions articulaires structurelles RA-III ont été évaluées radiographiquement et exprimées en changement du score net total (TSS) et de ses composants le score d'érosion et le score de rétrécissement des espaces conjoints (JSN) au mois 12 par rapport à la ligne de base. Au départ, le TSS médian était d'environ 55 dans le placebo et 40 mg toutes les deux groupes de la semaine. Les résultats sont présentés dans le tableau 6. Les patients traités par Humira / MTX ont démontré une progression moins radiographique que les patients recevant du MTX seul à 52 semaines.
Tableau 6: Changements moyens radiographiques sur 12 mois dans l'étude RA-III
| Placebo/ Mtx | / Mtx 40 mg toutes les deux semaines | Placebo/ Mtx-/ Mtx (95% Confidence Interval*) | P-Value ** | |
| Score pointu total | 2.7 | 0.1 | 2.6 (NULL,4 3,8) | <0.001 |
| Score d'érosion | 1.6 | 0.0 | 1,6 (NULL,9 2,2) | <0.001 |
| Score JSN | 1.0 | 0.1 | 0,9 (NULL,3 1,4) | 0.002 |
| * Intervalles de confiance à 95% pour les différences de scores de changement entre MTX et Humira. ** Basé sur l'analyse de rang |
Dans l'extension ouverte de l'étude RA-III, 77% des patients originaux traités avec une dose de Humira ont été évalués radiographiquement à 2 ans. Les patients ont maintenu l'inhibition des dommages structurels mesurés par le TSS. Cinquante-quatre pour cent n'avaient aucune progression des dommages structurels tels que définis par un changement dans le TSS de zéro ou moins. Cinquante-cinq pour cent (55%) des patients traités à l'origine avec 40 mg Humira toutes les deux semaines ont été évalués radiographiquement à 5 ans. Les patients ont eu une inhibition continue des dommages structurels avec 50% ne montrant aucune progression des dommages structurels définis par un changement dans le TSS de zéro ou moins.
Dans l'étude, les lésions articulaires structurelles RA-V ont été évaluées comme dans l'étude RA-III. Une plus grande inhibition de la progression radiographique évaluée par les changements dans le score d'érosion du TSS et le JSN a été observée dans le groupe de combinaison Humira / MTX par rapport au groupe de monothérapie MTX ou Humira à la semaine 52 ainsi qu'à la semaine 104 (voir le tableau 7).
Tableau 7: Changement moyen radiographique * dans l'étude RA-V
| MTX a N = 257 | AB N = 274 | / Mtx N = 268 | ||
| 52 semaines | Score pointu total | 5.7 (4.2 7.3) | 3.0 (NULL,7 4.3) | 1,3 (NULL,5 2,1) |
| Score d'érosion | 3.7 (NULL,7 4.8) | 1.7 (NULL,0 2,4) | 0,8 (NULL,4 1,2) | |
| Score JSN | 2.0 (NULL,2 2,8) | 1,3 (NULL,5 2,1) | 0,5 (NULL,0 1,0) | |
| 104 semaines | Score pointu total | 10.4 (NULL,7 13.2) | 5.5 (NULL,6 7,4) | 1,9 (NULL,9 2,9) |
| Score d'érosion | 6.4 (4.6 8.2) | 3.0 (2.0 4.0) | 1,0 (NULL,4 1,6) | |
| Score JSN | 4.1 (2.7 5.4) | 2.6 (NULL,5 3,7) | 0,9 (NULL,3 1,5) | |
| * moyenne (intervalle de confiance à 95%) a p <0.001 / Mtx vs. MTX at 52 et 104 weeks et for / Mtx vs. at 104 weeks b p <0.01 for / Mtx vs. at 52 semaines |
Réponse de la fonction physique
Dans les études, Ra-I via IV Humira a montré une amélioration significativement plus importante que le placebo dans le questionnaire sur l'indice des handicaps de l'évaluation de la santé (HAQ-DI) de la ligne de base à la fin de l'étude et une amélioration significativement plus importante que le placebo dans les résultats de la santé évalués par l'enquête sur la santé courte (SF 36). L'amélioration a été observée à la fois dans le résumé des composants physiques (PCS) et le résumé des composants mentaux (MCS).
Dans l'étude RA-III, l'amélioration moyenne (IC à 95%) de HAQ-Di par rapport à la semaine 52 était de 0,60 (NULL,55 0,65) pour les patients Humira et 0,25 (NULL,17 0,33) pour le placebo / mtx (P (P <0.001) patients. Sixty-three percent of -treated patients achieved a 0.5 or greater improvement in HAQ-DI at week 52 in the double-blind portion of the study. Eighty-two percent of these patients maintained that improvement through week 104 et a similar proportion of patients maintained this response through week 260 (5 years) of open-lABel treatment. Mean improvement in the SF-36 was maintained through the end of measurement at week 156 (3 years).
Dans l'étude RA-V, le HAQ-DI et la composante physique du SF-36 ont montré une amélioration plus importante (P <0.001) for the / Mtx combination therapy group versus either the MTX mopasherapy or the mopasherapy group at Semaine 52 which was maintained through Semaine 104.
Arthrite idiopathique juvénile
L'innocuité et l'efficacité de Humira ont été évaluées dans deux études (étudie Jia-I et Jia-II) chez les patients atteints d'arthrite idiopathique juvénile polyarticulaire active (JIA).
Étudier Jia-i
L'innocuité et l'efficacité de Humira ont été évaluées dans une étude de groupe parallèle en double aveugle randomisé multicentrique chez 171 patients âgés de 4 à 17 ans avec JIA polyarticulaire. Dans l'étude, les patients ont été stratifiés en deux groupes: traités par MTX ou non traités par MTX. Tous les patients ont dû montrer des signes de maladie modérée ou grave active malgré un traitement précédent avec des corticostéroïdes analgésiques AINS ou des ARM. Les patients qui ont reçu un traitement antérieur avec des ARM biologiques ont été exclus de l'étude.
L'étude a inclus quatre phases: un fil ouvert en phase (OL-LI; 16 semaines) une phase de retrait randomisée en double aveugle (dB; 32 semaines) une phase d'extension ouverte (OLE-BSA; jusqu'à 136 semaines) et une phase de dose fixe ouverte (OLE-FD; 16 semaines). Dans les trois premières phases de l'étude, Humira a été administré en fonction de la surface corporelle à une dose de 24 mg / m² jusqu'à une dose totale maximale de 40 mg par voie sous-cutanée (SC) toutes les deux semaines. Dans la phase OLE-FD, les patients ont été traités avec 20 mg de Humira SC toutes les deux semaines si leur poids était inférieur à 30 kg et avec 40 mg de Humira SC toutes les deux semaines si leur poids était de 30 kg ou plus. Les patients sont restés à des doses stables d'AINS et ou de prednisone (≤0,2 mg / kg / jour ou 10 mg / jour maximum).
Les patients démontrant une réponse pédiatrique ACR 30 à la fin de la phase OL-LI ont été randomisés dans la phase à double aveugle (dB) de l'étude et ont reçu Humira ou un placebo toutes les deux semaines pendant 32 semaines ou jusqu'à ce que la maladie flare. La poussée de la maladie a été définie comme une aggravation de ≥30% par rapport à la ligne de base dans ≥3 de 6 critères de base de l'ACR pédiatrique ≥ 2 articulations actives et une amélioration de> 30% dans un plus de 1 des 6 critères. Après 32 semaines ou au moment de la maladie de la maladie pendant la phase DB, les patients ont été traités dans la phase d'extension ouverte en fonction du régime BSA (Olebsa) avant de se convertir en un régime à dose fixe basé sur le poids corporel (phase Ole-FD).
Étudier Jia-i Réponse clinique
À la fin de la phase OL-LI de 16 semaines, 94% des patients de la strate MTX et 74% des patients de la strate non-MTX étaient des répondants pédiatriques ACR 30. Dans la phase DB, significativement moins de patients qui ont reçu Humira ont connu une poussée de maladie par rapport au placebo à la fois sans MTX (43% contre 71%) et avec MTX (37% contre 65%). Plus de patients traités par Humira ont continué à montrer des réponses pédiatriques ACR 30/50/70 à la semaine 48 par rapport aux patients traités par placebo. Les réponses pédiatriques ACR ont été maintenues jusqu'à deux ans dans la phase OLE chez les patients qui ont reçu Humira tout au long de l'étude.
Étudier Jia-iI
was assessed in an open-lABel multicenter study in 32 patients who were 2 to <4 years of age or 4 years of age et older weighing <15 kg with moderately to severely active polyarticular JIA. Most patients (97%) received at least 24 semaines of treatment dosed 24 mg/m² up to a maximum of 20 mg toutes les deux semaines as a single SC injection up to a maximum of 120 weeks duration. During the study most patients used concomitant MTX with fewer reporting use of corticosteroids or NSAIDs. The primary objective of the study was evaluation of safety [voir Effets indésirables ].
Arthrite psoriatique
L'innocuité et l'efficacité de Humira ont été évaluées dans deux études randomisées contrôlées par un placebo en double aveugle chez 413 patients atteints d'arthrite psoriasique (PSA). À la fin des deux études, 383 patients inscrits à une étude d'extension en plein air dans laquelle 40 mg Humira ont été administrés toutes les deux semaines.
L'étude PSA-I a inscrit 313 patients adultes avec un PSA modérément à sévère (> 3 articulations gonflées et> 3) qui ont eu une réponse inadéquate au traitement ANSA dans l'une des formes suivantes: (1) l'implication distale interphalangienne (DIP) (n = 23); (2) arthrite polyarticulaire (absence de nodules rhumatoïdes et présence de psoriasis en plaque) (n = 210); (3) arthrite mutilans (n = 1); (4) PSA asymétrique (n = 77); ou (5) en forme de type (n = 2). Les patients sous traitement MTX (158 des 313 patients) lors de l'inscription (dose stable de ≤ 30 mg / semaine pendant> 1 mois) pourraient continuer le MTX à la même dose. Des doses de Humira 40 mg ou d'un placebo toutes les deux semaines ont été administrées pendant la période en double aveugle de 24 semaines de l'étude.
Par rapport au traitement placebo avec Humira a entraîné une amélioration des mesures de l'activité de la maladie (voir les tableaux 8 et 9). Parmi les patients atteints de PSA qui ont reçu Humira, les réponses cliniques étaient apparentes chez certains patients au moment de la première visite (deux semaines) et ont été maintenues jusqu'à 88 semaines dans l'étude en cours en cours. Des réponses similaires ont été observées chez les patients atteints de chacun des sous-types de l'arthrite psoriasique, bien que peu de patients aient été inscrits avec l'arthrite mutilans et les sous-types de type spondylite ankylosante. Les réponses étaient similaires chez les patients qui recevaient ou ne recevaient pas une thérapie MTX concomitante au départ.
Les patients présentant une atteinte psoriasique d'une surface corporelle d'au moins trois pour cent (BSA) ont été évalués pour les réponses de la zone psoriatique et de l'indice de gravité (PASI). À 24 semaines, les proportions de patients atteignant une amélioration de 75% ou 90% du PASI étaient respectivement de 59% et 42% dans le groupe Humira (n = 69) contre 1% et 0% respectivement dans le groupe placebo (n = 69) (P <0.001). PASI responses were apparent in some patients at the time of the first visit (two weeks). Réponses were similar in patients who were or were pas receiving concomitant MTX therapy at baseline.
Tableau 8: Réponse de l'ACR dans l'étude PSA-I (pour cent des patients)
| Placebo N = 162 | * N = 151 | |
| Acr20 | ||
| Semaine 12 | 14% | 58% |
| Semaine 24 | 15% | 57% |
| Acr50 | ||
| Semaine 12 | 4% | 36% |
| Semaine 24 | 6% | 39% |
| ACR70 | ||
| Semaine 12 | 1% | 20% |
| Semaine 24 | 1% | 23% |
| * P <0.001 for all comparisons between et placebo |
Tableau 9: Composantes de l'activité de la maladie dans l'étude PSA-I
| Paramètre: médian | Placebo N = 162 | * N = 151 | ||
| Base de base | 24 semaines | Base de base | 24 semaines | |
| Nombre de joints tendres a | 23.0 | 17.0 | 20.0 | 5.0 |
| Nombre d'articulations gonflées b | 11.0 | 9.0 | 11.0 | 3.0 |
| Évaluation globale des médecins c | 53.0 | 49.0 | 55.0 | 16.0 |
| Évaluation globale des patients c | 49.5 | 49.0 | 48.0 | 20.0 |
| Douleur c | 49.0 | 49.0 | 54.0 | 20.0 |
| Indice de handicap (HAQ) d | 1.0 | 0.9 | 1.0 | 0.4 |
| CRP (Mg / DL) e | 0.8 | 0.7 | 0.8 | 0.2 |
| * P <0.001 for vs. placebo comparisons based on median changes a Échelle 0-78 b Échelle 0-76 c Échelle visuelle analogique; 0 = le meilleur 100 = pire d Indice de handicap du questionnaire sur l'évaluation de la santé; 0 = meilleur 3 = pire; Mesure la capacité du patient à effectuer les éléments suivants: Vobe / marié ARISE EAT WAKE REACH GRIP RETENU HYGIENE ET RETENU D'ACTIVITÉ TOUT. e Plage normale: 0-0.287 mg / dl |
Des résultats similaires ont été observés dans une étude supplémentaire de 12 semaines chez 100 patients atteints d'arthrite psoriasique modérée à sévère qui a eu une réponse sous-optimale au traitement par DMARD tel qu'il se manifeste par ≥3 articulations d'appel d'offres et ≥3 articulations gonflées lors de l'inscription.
Réponse radiographique
Les changements radiographiques ont été évalués dans les études PSA. Des radiographies des poignets et des pieds ont été obtenues au départ et à la semaine 24 pendant la période en double aveugle lorsque les patients étaient sur Humira ou un placebo et à la semaine 48 lorsque tous les patients étaient sous Humira en plein essor. Un score pointu total modifié (MTSS) qui comprenait des articulations interphalangiennes distales (c'est-à-dire non identiques au TSS utilisé pour la polyarthrite rhumatoïde) a été utilisée par les lecteurs aveugles à un groupe de traitement pour évaluer les radiographies.
-treated patients demonstrated greater inhibition of radiographic progression comparouge to placebo-treated patients et this effect was maintained at 48 semaines (see TABle 10).
Tableau 10: Modification du score net total modifié dans l'arthrite psoriasique
| Placebo N = 141 | N = 133 | ||
| Semaine 24 | Semaine 24 | Semaine 48 | |
| Base de base mean | 22.1 | 23.4 | 23.4 |
| Changement moyen ± ET | 0,9 ± 3,1 | -0,1 ± 1,7 | -0,2 ± 4,9 * |
| * <0.001 for the difference between Semaine 48 et Placebo Semaine 24 (primary analysis) |
Réponse de la fonction physique
Dans l'étude, la fonction physique du PSA-I et l'invalidité ont été évaluées à l'aide de l'indice de handicap HAQ (HAQ-DI) et de l'enquête sur la santé SF-36. Les patients traités avec 40 mg de Humira toutes les deux semaines ont montré une amélioration plus importante par rapport à la référence dans le score HAQ-DI (diminution moyenne de 47% et 49% aux semaines 12 et 24 respectivement) par rapport au placebo (diminution moyenne de 1% et 3% aux semaines 12 et 24 respectivement). Aux semaines 12 et 24, les patients traités par Humira ont montré une amélioration plus importante par rapport à la référence dans le score de résumé des composants physiques du SF-36 par rapport aux patients traités par placebo et sans empirement dans le score de résumé des composants mentaux SF-36. L'amélioration de la fonction physique basée sur le HAQ-DI a été maintenue jusqu'à 84 semaines grâce à la partie ouverte de l'étude.
Spondylarthrite ankylosante
L'innocuité et l'efficacité de Humira 40 mg toutes les deux semaines ont été évaluées chez 315 patients adultes dans une étude randomisée de 24 semaines à double aveugle contrôlée par un placebo chez des patients atteints de spondylarthrite ankylosante active (AS) qui a eu une réponse inadéquate aux glucocorticoïdes NSAIDS analgésiques méthotrexate ou sulfasalazine. Activé comme défini comme des patients qui remplissaient au moins deux des trois critères suivants: (1) un bain comme indice d'activité de la maladie (BASDAI) score ≥4 cm (2) un score visuel analogique (EVA) pour les maux de dos total ≥ 40 mm et (3) une rigidité matinale ≥ 1 heure. La période en aveugle a été suivie d'une période ouverte au cours de laquelle les patients ont reçu Humira 40 mg toutes les deux semaines par voie sous-cutanée jusqu'à 28 semaines supplémentaires.
L'amélioration des mesures de l'activité de la maladie a été observée pour la première fois à la semaine 2 et maintenue pendant 24 semaines comme le montre la figure 2 et le tableau 11.
Réponses of patients with total spinal ankylosis (n=11) were similar to those without total ankylosis.
Figure 2: Réponse ASAS 20 par visite Visite As-i
 |
À 12 semaines, les réponses ASAS 20/50/70 ont été obtenues de 58% 38% et 23% respectivement des patients recevant Humira contre 21% 10% et 5% respectivement de patients recevant un placebo (P <0.001). Similar responses were seen at Semaine 24 et were sustained in patients receiving open-lABel for up to 52 semaines.
Une plus grande proportion de patients traités avec Humira (22%) a atteint un faible niveau d'activité de la maladie à 24 semaines (définie comme une valeur <20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) comparouge to patients treated with placebo (6%).
Tableau 11: Composants de l'activité de la maladie de la spondylarthrite ankylosante
| Placebo N = 107 | N = 208 | |||
| Base de base mean | Semaine 24 mean | Base de base mean | Semaine 24 mean | |
| Ailes 20 critères Réponse * | ||||
| Évaluation globale du patient de l'activité de la maladie a * | 65 | 60 | 63 | 38 |
| Vaincu total du dos * | 67 | 58 | 65 | 37 |
| Inflammation b * | 6.7 | 5.6 | 6.7 | 3.6 |
| Basfi c * | 56 | 51 | 52 | 34 |
| Basdai d score* | 6.3 | 5.5 | 6.3 | 3.7 |
| Basmi e score* | 4.2 | 4.1 | 3.8 | 3.3 |
| Tragus au mur (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Flexion lombaire (CM) | 4.1 | 4.0 | 4.2 | 4.4 |
| Rotation cervicale (degrés) | 42.2 | 42.1 | 48.4 | 51.6 |
| Flexion côté lombaire (CM) | 8.9 | 9.0 | 9.7 | 11.7 |
| Distance intermalléolaire (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRP f * | 2.2 | 2.0 | 1.8 | 0.6 |
| a Pourcentage de sujets avec au moins une amélioration de 20% et 10 unités mesurée sur un visuel Échelle analogique (EVA) avec 0 = aucun et 100 = sévère b Moyenne des questions 5 et 6 de Basdai (définie dans «d ») c Bath ankylosing spondylart index fonctionnel d Indice d'activité de la maladie de la spondylarthrite ankylosante du bain e Indice de métrologie de la spondylite ankylosante du bain f Protéine C-réactive (Mg / DL) * Statistiquement significatif pour les comparaisons entre Humira et Placebo à la semaine 24 |
Une deuxième étude contrôlée par placebo en double aveugle randomisée de 82 patients atteints de spondylarthrite ankylosante a montré des résultats similaires.
Les patients traités par Humira ont obtenu une amélioration par rapport à la ligne de base du score du questionnaire sur la qualité de vie de la spondylarthrite ankylosante (ASQOL) (-3,6 vs -1,1) et dans le score de l'étude de la santé (SF-36) (SF-36) Score de composants (PCS) (7.4 contre 1,9) par rapport aux patients traités par placebo à la semaine 24.
Maladie adulte de Crohn
L'innocuité et l'efficacité de plusieurs doses de Humira ont été évaluées chez des patients adultes atteints de CD de la maladie de Crohn (Crohn (CDAI) de la maladie de Crohn (CDAI) dans les études randomisées en double aveugle contrôlée par le placebo en double aveugle (CDAI). Des doses stables concomitantes de corticostéroïdes aminosalicylates et / ou d'agents immunomodulatrices ont été autorisées et 79% des patients ont continué à recevoir au moins un de ces médicaments.
Induction de la rémission clinique (définie comme CDAI <150) was evaluated in two studies. In Study CD-I 299 TNF-blocker naïve patients were retomized to one of four treatment groups: the placebo group received placebo at Weeks 0 et 2 the 160/80 group received 160 mg at Week 0 et 80 mg at Week 2 the 80/40 group received 80 mg at Week 0 et 40 mg at Week 2 et the 40/20 group received 40 mg at Week 0 et 20 mg at Week 2. Clinical results were assessed at Semaine 4.
Dans la deuxième étude d'induction, CD-II 325 patients qui avaient perdu une réponse ou étaient intolérants à la thérapie infliximabe précédente ont été randomisés pour recevoir 160 mg Humira à la semaine 0 et 80 mg à la semaine 2 ou au placebo aux semaines 0 et 2. Les résultats cliniques ont été évalués à la semaine 4.
Le maintien de la rémission clinique a été évalué dans l'étude CD-III. Dans cette étude, 854 patients atteints d'une maladie active ont reçu un humira ouvert à 80 mg à la semaine 0 et 40 mg à la semaine 2. Les patients ont ensuite été randomisés à la semaine 4 à 40 mg Humira toutes les deux semaines 40 mg Humira chaque semaine ou placebo. La durée totale de l'étude était de 56 semaines. Les patients en réponse clinique (diminution du CDAI ≥70) à la semaine 4 ont été stratifiés et analysés séparément de ceux qui ne sont pas en réponse clinique à la semaine 4.
Induction de la rémission clinique
Un pourcentage plus élevé des patients traités avec 160/80 mg Humira ont obtenu l'induction de la rémission clinique par rapport au placebo à la semaine 4, que les patients soient un bloqueur de TNF Naã¯ve (CD-I) ou avaient perdu une réponse ou étaient intolérants à l'infliximab (CD-II) (voir tableau 12).
Tableau 12: Induction de la rémission clinique dans les études CD-I et CD-II (pour cent des patients)
| CD-I | CD-II | |||
| Placebo N = 74 | 160/80 mg N = 76 | Placebo N = 166 | 160/80 mg N = 159 | |
| Semaine 4 | ||||
| Rémission clinique | 12% | 36%* | 7% | 21% * |
| Réponse clinique | 34% | 58%** | 34% | 52% ** |
| Rémission clinique is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. * P <0.001 for vs. placebo pairwise comparison of proportions ** P <0.01 for vs. placebo pairwise comparison of proportions |
Entretien de la rémission clinique
Dans l'étude CD-III à la semaine 4, 58% (499/854) des patients étaient en réponse clinique et ont été évalués dans l'analyse primaire. Aux semaines 26 et 56, des proportions plus importantes de patients qui étaient en réponse clinique à la semaine 4 ont réalisé une rémission clinique dans le groupe d'entretien de 40 mg Humira toutes les deux semaines par rapport aux patients du groupe d'entretien placebo (voir tableau 13). Le groupe qui a reçu une thérapie Humira chaque semaine n'a pas démontré de taux de rémission significativement plus élevés par rapport au groupe qui a reçu Humira toutes les deux semaines.
Tableau 13: Maintien de la rémission clinique dans le CD-III (pour cent des patients)
| Placebo N = 170 | 40 mg humira toutes les deux semaines N = 172 | |
| Semaine 26 | ||
| Rémission clinique | 17% | 40% * |
| Réponse clinique | 28% | 54%* |
| Semaine 56 | ||
| Rémission clinique | 12% | 36%* |
| Réponse clinique | 18% | 43%* |
| Rémission clinique is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. * P <0.001 for vs. placebo pairwise comparisons of proportions |
Parmi ceux-ci en réponse à la semaine 4 qui ont atteint la rémission au cours de l'étude, les patients du Humira toutes les deux semaines ont maintenu la rémission pendant une période plus longue que les patients du groupe d'entretien placebo. Parmi les patients qui n'étaient pas en réponse à la semaine 12, le traitement s'est poursuivi au-delà de 12 semaines n'a pas entraîné de réponses significativement plus.
Maladie pédiatrique de Crohn
Une étude clinique randomisée en double aveugle de 52 semaines de 2 concentrations de dose de Humira (étude PCD-I) a été menée chez 192 patients pédiatriques (6 à 17 ans) avec une maladie de Crohn modérément à gravement active (défini le score de la maladie de Crohn de Crohn (PCDAI). Les patients inscrits ont eu au cours de la période précédente de deux ans une réponse inadéquate aux corticostéroïdes ou à un immunomodulateur (c'est-à-dire l'azathioprine 6-mercaptopurine ou le méthotrexate). Les patients qui avaient déjà reçu un bloqueur TNF ont été autorisés à s'inscrire s'ils avaient déjà eu une perte de réponse ou d'intolérance à ce bloqueur TNF.
Les patients ont reçu une thérapie d'induction ouverte à une dose en fonction de leur poids corporel (≥40 kg et <40 kg). Patients weighing ≥40 kg received 160 mg (at Week 0) et 80 mg (at Week 2). Patients weighing <40 kg received 80 mg (at Week 0) et 40 mg (at Week 2). At Semaine 4 patients within each body weight category (≥40 kg et <40 kg) were retomized 1:1 to one of two maintenance dose regimens (high dose et low dose). The high dose was 40 mg toutes les deux semaines for patients weighing ≥40 kg et 20 mg toutes les deux semaines for patients weighing <40 kg. The low dose was 20 mg toutes les deux semaines for patients weighing ≥40 kg et 10 mg toutes les deux semaines for patients weighing <40 kg.
Les doses stables concomitantes de corticostéroïdes (dosage de prednisone ≤40 mg / jour ou équivalent) et les immunomodulateurs (azathioprine 6-mercaptopurine ou méthotrexate) ont été autorisés tout au long de l'étude.
À la semaine 12, les patients qui ont connu une poussée de maladie (augmentation du PCDAI ≥ 15 à partir de la semaine 4 et du PCDAI absolu> 30) ou qui n'étaient pas des répondants (n'ont pas atteint une diminution du PCDAI ≥ 15 par rapport à 2 visites consécutives au moins 2 semaines d'intervalle) ont été autorisés à dose-escalate (c'est-à-dire à l'échange de toutes les autres semaines de dosting de l'abri de chaque semaine); Les patients qui ont été escaladés par la dose ont été considérés comme des échecs de traitement.
Au départ, 38% des patients recevaient des corticostéroïdes et 62% des patients recevaient un immunomodulateur. Quarante-quatre pour cent (44%) des patients avaient déjà perdu une réponse ou étaient intolérants à un bloqueur TNF. Le score PCDAI de base médian était de 40.
Sur les 192 patients au total, 188 patients ont terminé la période d'induction de 4 semaines, 152 patients ont effectué 26 semaines de traitement et 124 patients ont terminé 52 semaines de traitement. Cinquante et un pour cent (51%) (48/95) des patients dans la dose de dose à faible entretien écuniée et 38% (35/93) des patients dans la dose de dose à forte dose à forte dose.
À la semaine 4, 28% (52/188) des patients étaient en rémission clinique (définie comme PCDAI ≤ 10).
Les proportions de patients en rémission clinique (définie comme PCDAI ≤ 10) et la réponse clinique (définie comme une réduction du PCDAI d'au moins 15 points de la ligne de base) ont été évaluées aux semaines 26 et 52.
Aux semaines 26 et 52, la proportion de patients en rémission clinique et en réponse clinique était numériquement plus élevée dans le groupe à forte dose par rapport au groupe à faible dose (tableau 14). Le régime de maintenance recommandé est de 20 mg toutes les deux semaines pour les patients pesant <40 kg et 40 mg toutes les deux semaines for patients weighing ≥ 40 kg. Every week dosing is pas the recommended maintenance dosing regimen [voir Posologie et administration ].
Tableau 14: Rémission clinique et réponse clinique dans l'étude PCD-I
| Dose à faible entretien † (20 ou 10 mg toutes les deux semaines) N = 95 | Dose de maintenance élevée N = 93 | |
| Semaine 26 | ||
| Rémission clinique ‡ | 28% | 39% |
| Réponse clinique§ | 48% | 59% |
| Semaine 52 | ||
| Rémission clinique ‡ | 23% | 33% |
| Réponse clinique§ | 28% | 42% |
| † La dose à faible entretien était de 20 mg toutes les deux semaines pour les patients pesant ≥ 40 kg et 10 mg toutes les deux semaines pour les patients <40 kg. <40 kg. ‡ Rémission clinique définie comme PCDAI ≤ 10. § Réponse clinique définie comme une réduction du PCDAI d'au moins 15 points de la ligne de base. |
Colite ulcéreuse adulte
La sécurité et l'efficacité de Humira ont été évaluées chez des patients adultes atteints de colite ulcéreuse modérément à sévèrement active (score de mayo de 6 à 12 sur une échelle de 12 points avec un sous-ensemble d'endoscopie de 2 à 3 sur une échelle de 0 à 3) malgré un traitement simultané ou antérieur Uc-ii). Les deux études ont inscrit les patients naïques de blocage TNF, mais l'étude UC-II a également permis l'entrée de patients qui ont perdu la réponse ou étaient intolérants aux TNFBlockers. Quarante pour cent (40%) des patients inscrits à l'étude UC-II avaient déjà utilisé un autre bloqueur TNF.
Des doses stables concomitantes d'aminosalicylates et d'immunosuppresseurs ont été autorisées. Dans les études, les patients UC-I et II recevaient des corticostéroïdes aminosalicylates (69%) (59%) et / ou de l'azathioprine ou 6-MP (37%) au départ. Dans les deux études, 92% des patients ont reçu au moins un de ces médicaments.
L'induction de la rémission clinique (définie comme le score de mayo ≤ 2 sans sous-scores individuels> 1) à la semaine 8 a été évaluée dans les deux études. La rémission clinique à la semaine 52 et la rémission clinique soutenue (définie comme la rémission clinique aux deux semaines 8 et 52) ont été évaluées dans l'étude UC-II.
Dans l'étude, UC-I 390 Les patients atteints de blocs de TNF ont été randomisés dans l'un des trois groupes de traitement pour l'analyse d'efficacité primaire. Le groupe placebo a reçu un placebo aux semaines 0 2 4 et 6. Le groupe 160/80 a reçu 160 mg Humira à la semaine 0 et 80 mg à la semaine 2 et le groupe 80/40 a reçu 80 mg Humira à la semaine 0 et 40 mg à la semaine 2. Après la semaine 2, les patients des deux groupes de traitement Humira ont reçu 40 mg toutes les deux semaines.
Dans l'étude, les patients UC-II 518 ont été randomisés pour recevoir Humira 160 mg à la semaine 0 80 mg à la semaine 2 et 40 mg toutes les deux semaines à partir de la semaine 4 à la semaine 50 ou du placebo à partir de la semaine 0 et de toute la semaine à la semaine 50. Corticostéroïde a été autorisé à commencer à la semaine 8.
Dans les deux études UC-I et UC-II, un pourcentage plus élevé des patients traités avec 160/80 mg de Humira par rapport aux patients traités par placebo a obtenu une induction de rémission clinique. Dans l'étude UC-II, un pourcentage plus élevé des patients traités avec 160/80 mg de Humira par rapport aux patients traités par placebo a obtenu une rémission clinique soutenue (rémission clinique aux deux semaines 8 et 52) (tableau 15).
Tableau 15: Induction de la rémission clinique dans les études UC-I et UC-II et rémission clinique soutenue dans l'étude UC-II (pour cent des patients)
| Étude UC-I | Étude UC-II | |||||
| Placebo N = 130 | 160/80 mg N = 130 | Différence de traitement (IC à 95%) | Placebo N = 246 | 160/80 mg N = 248 | Différence de traitement (IC à 95%) | |
| Induction de la rémission clinique (rémission clinique à la semaine 8) | 9,2% | 18,5% | 9,3% * (NULL,9% 17,6%) | 9,3% | 16,5% | 7,2% * (NULL,2% 12,9%) |
| Rémission clinique soutenue (rémission clinique aux deux semaines 8 et 52) | N / A | N / A | N / A | 4,1% | 8,5% | 4,4% * (NULL,1% 8,6%) |
| La rémission clinique est définie comme un score de mayo ≤ 2 sans sous-scores individuels> 1. CI = intervalle de confiance * P <0.05 for vs. placebo pairwise comparison of proportions |
Dans l'étude UC-I, il n'y avait pas de différence statistiquement significative dans la rémission clinique observée entre le groupe Humira 80/40 mg et le groupe placebo à la semaine 8.
Dans l'étude UC-II, 17,3% (43/248) dans le groupe Humira étaient en rémission clinique à la semaine 52, contre 8,5% (21/246) dans le groupe placebo (différence de traitement: 8,8%; intervalle de confiance à 95% (IC): [2,8% 14,5%]; P <0.05).
Dans le sous-groupe de patients en étude, UC-II avec un blocage TNF préalable utilise la différence de traitement pour l'induction de la rémission clinique semble être inférieure à celle observée dans l'ensemble de la population d'étude et les différences de traitement pour la rémission clinique et la rémission clinique soutenue à la semaine 52 semblaient similaires à celles observées dans l'ensemble de la population d'étude. Le sous-groupe de patients avec une utilisation antérieure de blocage du TNF a atteint l'induction de la rémission clinique à 9% (9/98) dans le groupe Humira contre 7% (7/101) dans le groupe placebo et une rémission clinique soutenue à 5% (5/98) dans le groupe Humira contre 1% (1/101) dans le groupe placebo. Dans le sous-groupe de patients atteints de blocs de TNF antérieurs, l'utilisation de 10% (10/98) était en rémission clinique à la semaine 52 dans le groupe Humira contre 3% (3/101) dans le groupe placebo.
Colite ulcéreuse pédiatrique
La sécurité et l'efficacité de Humira ont été évaluées dans un essai en double aveugle randomisé multicentrique (étude PUC-I NCT02065557) chez 93 patients pédiatriques de 5 ans à 17 ans avec une colite ulcéreuse modérément à 3 points (un score de mayo corticostéroïdes et / ou immunomodulateur (c'est-à-dire l'azathioprine 6mercaptopurine ou méthotrexate). Quinze sur 93 patients (16%) dans l'étude avaient une expérience antérieure avec un bloqueur TNF. Les patients qui ont reçu des corticostéroïdes lors de l'inscription ont été autorisés à réduire leur corticothérapie après la semaine 4.
Soixante-dix-sept patients ont été initialement randomisés 3: 2 pour recevoir un traitement en double aveugle avec l'une des deux dosages de Humira. Les patients des deux groupes posologiques ont reçu 2,4 mg / kg (maximum de 160 mg) à la semaine 0 1,2 mg / kg (maximum de 80 mg) à la semaine 2 et 0,6 mg / kg (maximum de 40 mg) aux semaines 4 et 6. Le groupe de dosage plus élevé a également reçu une dosage supplémentaire de 2,4 mg / kg (maximum de 160 mg) à la semaine 1. Après la conception d'étude de 160 mg) à la semaine 1. Après AND pour la conception de 160 mg) à la semaine 1. Après ANNEMENT pour la conception de 160 mg) à la semaine 1. Après AND pour la conception de 160 mg) à la semaine 1. Après AND pour la conception de 160 mg) à la semaine 1. Après ANNEMENT pour la conception de 160 mg) à la semaine 1. Après ANNEMENT pour la conception de 160 mg) à la semaine 1. Après AND pour la conception d'étude 16. a reçu un traitement ouvert avec Humira à la dose supérieure.
At Week 8 62 patients who demonstrated clinical response per Partial Mayo Score (PMS; a subset of the Mayo score with no endoscopic component and defined as a decrease in PMS ≥ 2 points and ≥ 30% from baseline) were randomized equally to receive double-blind treatment with HUMIRA 0.6 mg/kg (maximum of 40 mg) every other week (lower dosage group) or 0.6 mg/kg (maximum of 40 mg) every Semaine (groupe à dosages plus élevé). Avant une modification de la conception de l'étude, 12 patients supplémentaires qui ont démontré une réponse clinique par PMS ont été randomisés pour recevoir un placebo.
Il n'y a aucune différence d'efficacité cliniquement pertinente anticipée entre la dose plus élevée étudiée au cours de l'essai PUC-I de 52 semaines et la posologie recommandée de Humira [voir Posologie et administration Pharmacologie clinique ].
Les patients qui répondaient aux critères de maladie de la maladie à la semaine 12 ou après ont été randomisés pour recevoir une dose de réinduction de 2,4 mg / kg (maximum de 160 mg) ou une dose de 0,6 mg / kg (maximum de 40 mg) puis ont poursuivi la dose à laquelle ils ont été randomisés à la semaine 8.
Les critères d'évaluation du co-primaire de l'étude étaient une rémission clinique par PMS (défini comme un PMS ≤ 2 et aucun sous-sol individuel> 1) à la semaine 8 et une rémission clinique par le score Mayo (défini comme un score de mayo ≤ 2 et aucun sous-sol individuel> 1) à la semaine 52 chez les patients qui ont atteint une réponse clinique par PMS à la semaine 8. 30% par rapport à la ligne de base) à la semaine 52 de la semaine 8 PMS Amélioration des répondeurs (définie comme un sous-sol de Mayo Endoscopy ≤ 1) à la semaine 52 de la semaine 8, les répondants et mayo marquent la rémission de la semaine 52 de la semaine 8 PMS.
Résultats de la semaine 8
À la semaine 8, la rémission du PMS a été obtenue de 60% [28/47; Intervalle de confiance à 95% (IC): (44% 74%)] des patients du groupe posologique plus élevé (sans compter les 16 patients recevant un dosage ouvert ouvert) et 43% [13/30; IC à 95%: (25% 63%)] des patients du groupe posologique inférieur. Les résultats du groupe de dosage supérieur sont représentatifs des résultats attendus avec la posologie recommandée [voir Posologie et administration Pharmacologie clinique ].
Semaine 52 Results
À la semaine 52, les critères d'évaluation ont été évalués dans la population de patients qui ont reçu un placebo en double aveugle 0,6 mg / kg (maximum de 40 mg) toutes les deux semaines ou Humira 0,6 mg / kg (maximum de 40 mg) chaque semaine entre la semaine 8 et la semaine 52 (tableau 16).
Tableau 16: Réponse clinique clinique et amélioration endoscopique à la semaine 52 chez les patients pédiatriques atteints de colite ulcéreuse (étude PUC-1)
| Placebo a N / N (%) 95% IC | Maximum of 40 mg (0.6 mg/kg) every other week b N / N (%) 95% IC | Maximum of 40 mg (0.6 mg/kg) every week c N / N (%) 95% IC | |
| Rémission clinique in Week 8 PMS responders | 4/12 (33%) (10% 65%) | 9/31 (29%) (14% 48%) | 14/31 (45%) (27% 64%) |
| Réponse clinique in Week 8 PMS responders | 4/12 (33%) (10% 65%) | 19/31 (61%) (42% 78%) | 21/31 (68%) (49% 83%) |
| Amélioration endoscopique de la semaine 8 répondants PMS | 4/12 (33%) (10% 65%) | 12/31 (39%) (22% 58%) | 16/31 (52%) (33% 70%) |
| Rémission clinique in Week 8 PMS remitters | 3/8 (38%) (9% 76%) | 9/21 (43%) (22% 66%) | 10/22 (45%) (24% 68%) |
| CI = intervalle de confiance a Douze patients qui ont démontré une réponse clinique par PM à la semaine 8 ont été randomisés pour recevoir un placebo. Il y a des limites à l'interprétabilité des données du placebo en raison de la petite taille de l'échantillon. b Le dosage de toutes les deux semaines étudié au cours de l'essai PUC-I de 52 semaines est une posologie plus faible que la dose recommandée de Humira [voir Posologie et administration ]. c Il n'y a aucune différence d'efficacité cliniquement pertinente anticipée entre la dose plus élevée étudiée au cours de l'essai PUC-I de 52 semaines et la dose recommandée de Humira. Remarque: Les patients présentant des valeurs manquants à la semaine 52 ou qui ont été randomisés pour recevoir un traitement de réinduction ou d'entretien en raison de la poussée de la maladie ont été considérés comme des non-répondants pour les critères finaux de la semaine 52. |
Psoriasis en plaques
L'innocuité et l'efficacité de Humira ont été évaluées dans des études randomisées en double aveugle en double aveugle dans 1696 sujets adultes atteints de psoriasis de plaque chronique modéré à sévère (PS) qui étaient candidats à une thérapie systémique ou à la photothérapie.
L'étude PS-I a évalué 1212 sujets atteints de PS chronique avec ≥ 10% de surface corporelle (BSA) Évaluation globale (PGA) de la gravité de la maladie et du psoriasis et de l'indice de gravité (PASI) ≥12 au moins dans trois périodes de traitement. Au cours de la période, les sujets ont reçu un placebo ou un humira à une dose initiale de 80 mg à la semaine 0 suivi d'une dose de 40 mg toutes les deux semaines à partir de la semaine 1. Après 16 semaines de sujets de thérapie qui ont atteint au moins une réponse PASI 75 à la semaine 16 définie comme une amélioration du score pasi 40 mg Humira au moins 75% par rapport à la semaine.
Après 17 semaines de sujets de thérapie à marque ouverte qui ont maintenu au moins une réponse PASI 75 à la semaine 33 et ont été à l'origine randomisées en thérapie active au cours de la période A ont été re-randomisées dans la période C pour recevoir 40 mg Humira toutes les deux semaines ou un placebo pendant 19 semaines supplémentaires. Dans tous les groupes de traitement, le score PASI de base moyen était de 19 et le score d'évaluation global du médecin de base variait de modéré (53%) à sévère (41%) à très sévère (6%).
L'étude PS-II a évalué 99 sujets randomisés à Humira et 48 sujets randomisés dans un placebo avec un psoriasis de plaque chronique avec une atteinte à ≥10% de BSA et PASI ≥12. Les sujets ont reçu un placebo ou une dose initiale de 80 mg Humira à la semaine 0 suivie de 40 mg toutes les deux semaines à partir de la semaine 1 pendant 16 semaines. Dans tous les groupes de traitement, le score PASI de base moyen était de 21 et le score PGA de base variait de modéré (41%) à sévère (51%) à très sévère (8%).
Les études PS-I et II ont évalué la proportion de sujets qui ont atteint une maladie claire ou minimale sur l'échelle de PGA à 6 points et la proportion de sujets qui ont atteint une réduction du score PASI d'au moins 75% (PASI 75) par rapport à la semaine 16 (voir tableau 17 et 18).
De plus, l'étude PS-I a évalué la proportion de sujets qui ont maintenu un PGA de maladie claire ou minimale ou une réponse PASI 75 après la semaine 33 et la semaine 52 ou avant.
Tableau 17: Résultats de l'efficacité à 16 semaines dans l'étude PS-I Nombre de sujets (%)
| 40 mg toutes les deux semaines N = 814 | Placebo N = 398 | |
| PGA: Clair ou minimal * | 506 (62%) | 17 (4%) |
| Après 75 | 578 (71%) | 26 (7%) |
| * Clear = pas d'élévation de la plaque pas d'échelle plus ou moins d'hyperpigmentation ou de coloration rose ou rouge diffuse minimale = possible mais difficile à déterminer s'il y a une légère élévation de la plaque au-dessus de la peau normale ou de la sécheresse de surface moins avec une coloration blanche plus ou moins jusqu'à la coloration rouge |
Tableau 18: Résultats de l'efficacité à 16 semaines dans l'étude PS-II Nombre de sujets (%)
| 40 mg toutes les deux semaines N = 99 | Placebo N = 48 | |
| PGA: Clair ou minimal * | 70 (71%) | 5 (10%) |
| Après 75 | 77 (78%) | 9 (19%) |
| * Clear = pas d'élévation de la plaque pas d'échelle plus ou moins d'hyperpigmentation ou de coloration rose ou rouge diffuse Minimal = possible mais difficile à déterminer s'il y a une légère élévation de la plaque au-dessus de la peau normale plus ou moins la sécheresse de surface avec une coloration blanche plus ou moins jusqu'à la coloration rouge |
De plus, dans l'étude, les sujets PS-I sur Humira qui ont maintenu un PASI 75 ont été ré-alés 75 (79% contre 43%).
Au total, 347 intervenants stables ont participé à une évaluation de retrait et de retraitement dans une étude d'extension en plein air. Le délai médian de rechute (diminuer à la PGA modérée ou pire) était d'environ 5 mois. Pendant la période de retrait, aucun sujet n'a subi de transformation en psoriasis pustuleux ou érythrodermique. Au total, 178 sujets qui ont rechuté un traitement réinitialisé avec 80 mg de Humira puis 40 mg toutes les deux semaines à partir de la semaine 1. À la semaine 16 69% (123/178) des sujets ont eu une réponse de PGA claire ou minimale.
Une étude en double aveugle randomisée (étude PS-III) a comparé l'efficacité et l'innocuité de Humira par rapport au placebo chez 217 sujets adultes. Les sujets de l'étude devaient avoir un psoriasis de plaque chronique d'au moins une gravité modérée sur l'échelle de l'ongle de l'ongle à l'échelle de la PGA d'une gravité au moins modérée sur une échelle de psoriasis de psoriasis de ongle de ongle (PGA-F) à 5 points, un indice de gravité de l'ongle modifié (MNAPSI) pour le score cible-finni 10% ou une implication de BSA d'au moins 5% avec un score MNAPSI total pour toutes les ongles ≥ 20. Les sujets ont reçu une dose initiale de 80 mg Humira suivie de 40 mg toutes les deux semaines (à partir d'une semaine après la dose initiale) ou un placebo pendant 26 semaines suivis d'un traitement HuMira ouvert pendant 26 semaines supplémentaires. Cette étude a évalué la proportion de sujets qui ont obtenu une évaluation claire ou minimale avec au moins une amélioration à 2 gras sur l'échelle PGA-F et la proportion de sujets qui ont atteint au moins une amélioration de 75% par rapport à la ligne de base dans le score MNAPSI (MNAPSI 75) à la semaine 26.
À la semaine 26, une proportion plus élevée de sujets dans le groupe Humira que dans le groupe placebo a atteint le point final PGA-F. De plus, une proportion plus élevée de sujets dans le groupe Humira que dans le groupe placebo a atteint le MNAPSI 75 à la semaine 26 (voir tableau 19).
Tableau 19: Résultats de l'efficacité à 26 semaines
| Point final | 40 mg toutes les deux semaines* N = 109 | Placebo N = 108 |
| PGA-F: amélioration ≥ 2 grades et clair ou minimal | 49% | 7% |
| MNAPS 75 | 47% | 3% |
| * Les sujets ont reçu 80 mg de Humira à la semaine 0 suivi de 40 mg toutes les deux semaines à partir de la semaine 1. |
La douleur aux ongles a également été évaluée et une amélioration de la douleur à l'ongle a été observée dans l'étude PS-III.
Hidradénite suppurative
Deux études randomisées contrôlées par placebo en double aveugle (études HS-I et II) ont évalué l'innocuité et l'efficacité de Humira chez un total de 633 sujets adultes atteints de la Hidradénite suppurativa modérée à sévère (HS) avec une maladie de stade II ou III et avec au moins 3 abcès ou nodules inflammatoires. Dans les deux études, les sujets ont reçu un placebo ou Humira à une dose initiale de 160 mg à la semaine 0 80 mg à la semaine 2 et 40 mg chaque semaine à partir de la semaine 4 et se sont poursuivies pendant la semaine 11. Les sujets ont utilisé quotidiennement un lavage antiseptique topique. L'utilisation concomitante des antibiotiques orales a été autorisée dans l'étude HS-II.
Les deux études ont évalué la réponse clinique de la Hidradénite suppurativa (HISCR) à la semaine 12. HISCR a été définie comme une réduction au moins de 50% du nombre total d'abcès et des nodules inflammatoires sans augmentation du nombre d'abcès et aucune augmentation du nombre de fistules de vidange par rapport à la base de base (voir le tableau 18). La réduction de la douleur cutanée liée au HS a été évaluée en utilisant une échelle de notation numérique chez les patients qui sont entrés dans l'étude avec un score de base initial de 3 ou plus sur une échelle de 11 points.
Dans les deux études, une proportion plus élevée de sujets Humira que traités par placebo a atteint HISCR (voir tableau 20).
Tableau 20: Résultats de l'efficacité à 12 semaines chez les sujets avec une idradénite modérée à sévère suppurativa
| Étude HS I | Étude HS II* | |||
| Placebo | Humira 40 mg Weekly | Placebo | Humira 40 mg Weekly | |
| Hidradénite suppurative Réponse clinique (HiSCR) | N = 154 40 (26%) | N = 153 64 (42%) | N = 163 45 (28%) | N = 163 96 (59%) |
| * 19,3% des sujets de l'étude HS-II ont continué l'antibiotique orale de base au cours de l'étude. |
Dans les deux études de la semaine 12 à la semaine 35 (période B), les sujets qui avaient reçu Humira ont été re-randomisés à 1 des 3 groupes de traitement (Humira 40 mg chaque semaine Humira 40 mg toutes les deux semaines ou placebo). Les sujets qui avaient été randomisés dans le placebo ont été chargés de recevoir Humira 40 mg chaque semaine (étude HS-I) ou placebo (étude HS-II).
Pendant la période, la rafale B de HS définie comme une augmentation ≥25% par rapport à la référence dans les abcès et le nombre de nodules inflammatoires et avec un minimum de 2 lésions supplémentaires a été documentée dans 22 (22%) des 100 sujets qui ont été retirés du traitement Humira après le trimestre d'efficacité primaire dans deux études.
Uvéite adulte
L'innocuité et l'efficacité de Humira ont été évaluées chez des patients adultes atteints d'intermédiaire non infectieux et de panuvéite intermédiaire excluant les patients atteints d'uvéite antérieure isolée dans deux études contrôlées par placebo à double masquage randomisées (UV I et II). Les patients ont reçu un placebo ou un humira à une dose initiale de 80 mg suivie de 40 mg toutes les deux semaines à partir d'une semaine après la dose initiale. Le critère d'évaluation de l'efficacité principale dans les deux études était le temps de l'échec du traitement ».
L'échec du traitement était un résultat multi-composantes défini comme le développement de nouvelles lésions vasculaires rétiniennes choriorétinales et / ou inflammatoires inflammatoires une augmentation de la qualité cellulaire de la chambre antérieure (AC) ou de la brume vitreuse (VH) ou une diminution de la meilleure acuité visuelle corrigée (BCVA).
L'étude UV I a évalué 217 patients atteints d'uvéite active tout en étant traités avec des corticostéroïdes (prednisone orale à une dose de 10 à 60 mg / jour). Tous les patients ont reçu une dose standardisée de prednisone 60 mg / jour à l'entrée de l'étude, suivie d'un calendrier conique obligatoire avec une interruption complète des corticostéroïdes d'ici la semaine 15.
L'étude UV II a évalué 226 patients atteints d'uvéite inactive tout en étant traités avec des corticostéroïdes (prednisone orale 10 à 35 mg / jour) au départ pour contrôler leur maladie. Les patients ont par la suite subi un calendrier de cône obligatoire avec une arrêt complet des corticostéroïdes d'ici la semaine 19.
Réponse clinique
Les résultats des deux études ont démontré une réduction statistiquement significative du risque d'échec du traitement chez les patients traités par Humira par rapport aux patients recevant un placebo. Dans les deux études, toutes les composantes du critère d'évaluation principal ont contribué cumulativement à la différence globale entre les groupes Humira et placebo (tableau 21).
Tableau 21: Temps de l'échec du traitement dans les études UV I et UV II
| UV I | UV II | |||||
| Placebo (N = 107) | (N = 110) | HR [95% IC] a | Placebo (N = 111) | (N = 115) | HR [95% IC] a | |
| Échec b n (%) | 84 (NULL,5) | 60 (NULL,5) | 0.50 [0,36 0,70] | 61 (55.0) | 45 (39.1) | 0.57 [0,39 0,84] |
| Délai médian à l'échec (mois) [IC à 95%] | 3.0 [2.7 3.7] | 5.6 [3.9 9.2] | N / A | 8.3 [4.8 12.0] | Ne c | N / A |
| a HR de Humira versus placebo à partir de la régression des risques proportionnels avec le traitement comme facteur. b L'échec du traitement à la semaine 6 ou après dans les UV d'étude I ou à ou après la semaine 2 dans l'étude UV II a été considéré comme un événement. Les sujets qui ont interrompu l'étude ont été censurés au moment de l'abandon. c Ne = pas estimABle. Fewer than half of at-risk subjects had an event. |
Figure 3: Les courbes de Kaplan-Meier résumant le temps à l'échec du traitement à la semaine 6 (étude UV I) ou semaine 2 (étude UV II)
 |
Remarque: P
Uvéite pédiatrique
L'innocuité et l'efficacité de Humira ont été évaluées dans une étude aléatoire randomisée à double masqué <18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Patients received either placebo or 20 mg adalimumab (if < 30 kg) or 40 mg adalimumab (if ≥ 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a metatory rougeuction in topical corticosteroids within 3 months.
Le critère d'évaluation principal était «le temps de l'échec du traitement». Les critères déterminant l'échec du traitement aggravaient ou non plus sans amélioration dans l'inflammation oculaire ou l'aggravation des comorbidités oculaires.
Réponse clinique
significantly decreased the risk of treatment failure by 75% relative to placebo (HR = 0.25 [95% CI: 0.12 0.49]) (TABle 22).
Tableau 22: Résultats de l'analyse du temps à l'échec du traitement (étude PUV-I)
| Placebo | (N = 60) | RH (IC à 95%) a | |
| Échec (n[%]) | 18 (60%) | 16 (NULL,7%) | 0,25 (NULL,12 0,49) |
| Délai médian à l'échec (semaines) (IC à 95%) b | 24.1 (12.4 81.0) | NeC | |
| a HR de l'adalimumab par rapport au placebo à partir de la régression des risques proportionnels avec le traitement comme facteur. b Estimé sur la base de la courbe de Kaplan-Meier. c Ne = pas estimABle. Fewer than half of at-risk subjects had an event. |
Figure 4: Courbes de Kaplan-Meier résumant le temps à l'échec du traitement (étude PUV-I)
 |
Étudier PUV-i
Remarque: p = placebo (nombre à risque); H = Humira (nombre à risque).
Références
1.National Cancer Institute. Programme de base de données sur les résultats (SEER) de l'épidémiologie et des résultats finaux de la surveillance. Incidence du SEER Tarifs du brut 17 registres 2000-2007.
Informations sur les patients pour Humira
®
(Hu-Mare-ah)
(adalimumab)
injection pour une utilisation sous-cutanée
Lisez le guide des médicaments qui est livré avec Humira avant de commencer à le prendre et chaque fois que vous obtenez une recharge. Il peut y avoir de nouvelles informations. Ce guide de médicaments ne prend pas la place de parler avec votre médecin de votre état de santé ou de votre traitement.
Quelles sont les informations les plus importantes que je devrais connaître sur Humira?
is a medicine that affects your immune system. can lower the ABility of your immune system to fight infections. Des infections graves se sont produites chez des gens qui prennent Humira. Ces infections graves comprennent la tuberculose (TB) et les infections causées par des champignons ou des bactéries des virus qui se sont propagés dans tout le corps. Certaines personnes sont mortes de ces infections.
- Votre médecin doit vous tester pour la tuberculose avant de commencer Humira.
- Votre médecin doit vous vérifier de près pour les signes et symptômes de la tuberculose pendant le traitement avec Humira.
Vous ne devriez pas commencer à prendre Humira si vous avez une sorte d'infection à moins que votre médecin ne dise que tout va bien.
Avant de commencer Humira, dites à votre médecin si vous:
- Pensez que vous avez une infection ou que vous avez des symptômes d'une infection tels que:
- La fièvre transpire ou frisson
- maux de muscle
- toux
- essoufflement
- sang dans le flegme
- Red chaud ou peau douloureuse ou plaies sur votre corps
- Diarrhée ou douleur à l'estomac
- brûler lorsque vous urinez ou urinez plus souvent que la normale
- se sentir très fatigué
- perte de poids
- sont traités pour une infection.
- Obtenez beaucoup d'infections ou ayant des infections qui continuent de revenir.
- avoir le diabète.
- avoir une tuberculose ou avoir été en contact étroit avec quelqu'un avec TB.
- sont nés dans des pays ou voyagés dans les pays où il y a plus de risque d'obtention de la tuberculose. Demandez à votre médecin si vous n'êtes pas sûr.
- vivent ou ont vécu dans certaines parties du pays (comme les vallées de la rivière Ohio et Mississippi) où il existe un risque accru d'obtenir certains types d'infections fongiques (histoplasmose coccidiomidomycose ou blastomycose). Ces infections peuvent se produire ou devenir plus graves si vous utilisez Humira. Demandez à votre médecin si vous ne savez pas si vous avez vécu dans une zone où ces infections sont courantes.
- ont ou ont eu l'hépatite B.
- Utilisez la médecine Orencia (Abatacept) Kineret (rituximab) ou purinethol (6-mercaptopurine 6-MP).
- sont prévus pour subir une chirurgie majeure.
Après avoir commencé Humira, appelez votre médecin tout de suite Si vous avez une infection ou tout signe d'une infection.
can make you more likely to get infections or make any infection that you may have worse.
Cancer
- Pour les enfants et les adultes qui prennent des bloqueurs de facteur de nécrose tumorale (TNF), y compris Humira, les chances d'obtenir un cancer peuvent augmenter.
- Il y a eu des cas de cancers inhabituels chez les enfants adolescents et les jeunes adultes utilisant des blocs TNF.
- Les personnes atteintes de polyarthrite rhumatoïde (PR), en particulier plus de PR grave, peuvent avoir plus de chances d'obtenir une sorte de cancer appelé lymphome.
- Si vous utilisez des bloqueurs TNF, y compris Humira, votre chance d'obtenir deux types de cancer de la peau peut augmenter (cancer des cellules basales et cancer squameux de la peau). Ces types de cancer ne sont généralement pas mortels s'ils sont traités. Dites à votre médecin si vous avez une bosse ou une plaie ouverte qui ne guérit pas.
- Certaines personnes recevant des bloqueurs TNF, dont Humira, ont développé un type de cancer rare appelé lymphome hépatosplénique à cellules T. Ce type de cancer entraîne souvent la mort. La plupart de ces personnes étaient des adolescents masculins ou des jeunes hommes. De plus, la plupart des gens étaient traités pour la maladie de Crohn ou la colite ulcéreuse avec un autre médicament appelé Imuran (azathioprine) ou purinethol (6-mercaptopurine 6 - MP).
Qu'est-ce que Humira?
is a medicine called a Tumor Necrosis Factor (TNF) blocker. is used:
- Pour réduire les signes et symptômes de:
- PR modérée à sévère chez les adultes. can be used alone with methotrexate or with certain other medicines.
- Arthrite idiopathique juvénile polyarticulaire modérée à sévère (JIA) chez les enfants 2 ans et plus. Humira peut être utilisé seul ou avec du méthotrexate.
- arthrite psoriatique (PsA) in adults. can be used alone or with certain other medicines.
- spondylarthrite ankylosante (AS) in adults.
- Hidradénite modérée à sévère suppurativa (HS) chez les personnes de 12 ans et plus.
- Pour traiter la maladie (CD) de Crohn modérée à sévère chez les adultes et les enfants de 6 ans et plus.
- Traiter la colite ulcéreuse modérée à sévère (UC) chez les adultes et les enfants de 5 ans et plus. On ne sait pas si Humira est efficace chez les personnes qui ont cessé de répondre ou ne pouvaient pas tolérer des médicaments à blocs de TNF.
- Pour traiter le psoriasis de plaque chronique (PS) modéré à sévère (durable) chez les adultes qui ont la condition dans de nombreuses régions de leur corps et qui peuvent bénéficier de la prise d'injections ou de pilules (thérapie systémique) ou de photothérapie (traitement en utilisant la lumière ultraviolette seule ou avec des pilules).
- Traiter l'intermédiaire non infectieux et la panuvéite intermédiaire chez les adultes et les enfants de 2 ans et plus.
Que dois-je dire à mon médecin avant de prendre Humira?
may pas be right for you. Before commencering tell your doctor ABout all of your medical conditions including if you:
- avoir une infection. Voir Quelles sont les informations les plus importantes que je devrais connaître sur Humira?
- ont ou ont eu un cancer.
- avoir un engourdissement ou des picotements ou avoir une maladie qui affecte votre système nerveux tel que sclérose en plaques ou syndrome de Guillain-Barré.
- avoir ou souffrir d'insuffisance cardiaque.
- ont récemment reçu ou devraient recevoir un vaccin. Vous pouvez recevoir des vaccins à l'exception des vaccins en direct tout en utilisant Humira. Les enfants doivent être mis à jour avec tous les vaccins avant de commencer Humira.
- sont allergiques au caoutchouc ou au latex. Dites à votre médecin si vous avez des allergies au caoutchouc ou au latex.
- La couverture d'aiguille pour le stylo Humira 40 mg / 0,8 ml Humira 40 mg / 0,8 ml de seringue pré-remplie Humira 20 mg / 0,4 ml de seringue préfabillée et Humira 10 mg / 0,2 ml La seringue préfabillée peut contenir du caoutchouc ou du latex naturel.
- La couverture d'aiguille noire pour le stylo Humira 80 mg / 0,8 ml Humira 80 mg / 0,8 ml Syringe préfabillé Humira Pen 40 mg / 0,4 ml Humira 40 mg / 0,4 ml fait avec du caoutchouc naturel ou du latex.
- sont allergiques à Humira ou à l'un de ses ingrédients. Voir la fin de ce guide de médicaments pour une liste d'ingrédients à Humira.
- sont enceintes ou envisagent de devenir enceintes d'allaitement enceintes ou prévoient d'allaiter. Vous et votre médecin devriez décider si vous devriez prendre Humira pendant que vous êtes enceinte ou allaitez.
- Ayez un bébé et vous utilisiez Humira pendant votre grossesse. Dites le médecin de votre bébé avant que votre bébé ne reçoive des vaccins.
Parlez à votre médecin de tous les médicaments que vous prenez y compris les médicaments sur ordonnance et les médicaments sur-thecounter et les suppléments à base de plantes.
Parlez surtout de votre médecin si vous utilisez:
- Orencia (Abatacept) Kineret (Anakinra) Remicade (Infliximab) Enbrel (Etanercept) Cimzia (certiolizumab Pegol) ou Simponi (golimumab) parce que vous ne devriez pas utiliser Humira pendant que vous utilisez également l'un de ces médicaments.
- Rituxan (rituximab). Votre médecin peut ne pas vouloir vous donner Humira si vous avez reçu Rituxan (rituximab) récemment.
- Imuran (azathioprine) ou purinethol (6-mercaptopurine 6-MP).
Gardez une liste de vos médicaments avec vous pour montrer à votre médecin et à votre pharmacien chaque fois que vous obtenez un nouveau médicament.
Comment devrais-je prendre Humira?
- is given by an injection under the skin. Your doctor will tell you how often to take an injection of . This is based on your condition to be treated. N'injectez pas Humira plus souvent que vous ne l'avez prescrit.
- Voir les instructions à utiliser à l'intérieur du carton pour des instructions complètes pour la bonne façon de préparer et d'injecter Humira.
- Assurez-vous que l'on vous a montré comment injecter Humira avant de le faire vous-même. Vous pouvez appeler votre médecin ou le 1-800-4Humira (1-800-448-6472) si vous avez des questions sur vous donner une injection. Quelqu'un que vous connaissez peut également vous aider avec votre injection après qu'il a été montré comment préparer et injecter Humira.
- Ne pas Essayez d'injecter vous-même Humira jusqu'à ce que vous ayez montré la bonne façon de donner les injections. Si votre médecin décide que vous ou un soignant peut être en mesure de donner vos injections d'Humira à la maison, vous devriez recevoir une formation sur la bonne façon de préparer et d'injecter Humira.
- Ne pas miss any doses of unless your doctor says it is okay. If you forget to take inject a dose as soon as you remember. Then take your next dose at your regular scheduled time. This will put you back on schedule. In case you are pas sure when to inject call your doctor or pharmacist.
- Si vous prenez plus Humira, on vous a dit de prendre votre médecin.
Quels sont les effets secondaires possibles de Humira?
can cause serious side effects including:
Vous voyez quelles sont les informations les plus importantes que je devrais connaître sur Humira?
- Infections graves.
Votre médecin vous examinera pour la tuberculose et effectuera un test pour voir si vous avez TB. Si votre médecin estime que vous êtes à risque de tuberculose, vous pouvez être traité avec des médicaments pour la tuberculose avant de commencer le traitement par Humira et pendant le traitement avec Humira. Même si votre Test de tuberculose est négatif, votre médecin doit vous surveiller attentivement pour les infections à la tuberculose pendant que vous prenez Humira. Les personnes qui ont passé un test de peau de TB négative avant de recevoir Humira ont développé une tuberculose active. Dites à votre médecin si vous avez l'un des symptômes suivants lors de la prise ou après la prise de Humira:
- toux that does pas go away
- Fièvre de bas grade
- perte de poids
- Perte de graisse corporelle et de muscle (gaspillage)
- Infection par l'hépatite B chez les personnes qui portent le virus dans leur sang.
Si vous êtes un porteur du virus de l'hépatite B (un virus qui affecte le foie), le virus peut devenir actif pendant que vous utilisez Humira. Votre médecin doit faire des tests sanguins avant de commencer le traitement pendant que vous utilisez Humira et pendant plusieurs mois après votre arrêt de traitement avec Humira. Dites à votre médecin si vous avez l'un des symptômes suivants d'une éventuelle infection à l'hépatite B:
- maux de muscle
- se sentir très fatigué
- urine sombre
- La peau ou les yeux ont l'air jaune
- peu ou pas d'appétit
- vomissement
- semelles colorées en argile
- fièvre
- frissons
- malaise
- éruption cutanée
- Réactions allergiques. Des réactions allergiques peuvent se produire chez les personnes qui utilisent Humira. Appelez votre médecin ou obtenez une aide médicale immédiatement si vous présentez l'un de ces symptômes d'une réaction allergique grave:
- urticaire
- difficulté à respirer
- gonflement de ton visage les yeux lèvres ou bouche
- Problèmes du système nerveux. Les signes et symptômes d'un problème de système nerveux comprennent: un engourdissement ou des problèmes de picotement avec votre faiblesse de la vision dans vos bras ou vos jambes et vos étourdissements.
- Problèmes de sang. Votre corps peut ne pas faire suffisamment de cellules sanguines qui aident à lutter contre les infections ou aident à arrêter les saignements. Les symptômes incluent une fièvre qui ne disparaît pas très facilement les ecchymoses ou les saignements ou l'air très pâle.
- Nouvelle insuffisance cardiaque ou aggravation de l'insuffisance cardiaque que vous avez déjà. Appelez votre médecin tout de suite Si vous obtenez de nouveaux symptômes d'aggravation d'insuffisance cardiaque tout en prenant Humira, notamment:
- essoufflement
- gain de poids soudain
- gonflement de vos chevilles ou pieds
- Réactions immunitaires, y compris un syndrome de type lupus. Les symptômes incluent l'inconfort thoracique ou la douleur qui ne disparaît pas contre l'essoufflement des douleurs articulaires ou une éruption cutanée sur vos joues ou vos bras qui empire au soleil. Les symptômes peuvent s'améliorer lorsque vous arrêtez Humira.
- Problèmes hépatiques. Des problèmes hépatiques peuvent se produire chez les personnes qui utilisent des médicaments contre le bloqueur TNF. Ces problèmes peuvent entraîner une insuffisance hépatique et une mort. Appelez votre médecin immédiatement si vous avez l'un de ces symptômes:
- se sentir très fatigué
- Mauvais appétit ou vomissements
- La peau ou les yeux ont l'air jaune
- Douleur sur le côté droit de votre estomac (abdomen)
- Psoriasis. Certaines personnes utilisant Humira avaient un nouveau psoriasis ou une aggravation du psoriasis qu'ils avaient déjà. Dites à votre médecin si vous développez des patchs écailleux rouges ou des bosses surélevées remplies de pus. Votre médecin peut décider d'arrêter votre traitement avec Humira.
Appelez votre médecin ou prenez immédiatement des soins médicaux si vous développez l'un des symptômes ci-dessus. Votre traitement avec Humira peut être arrêté.
Les effets secondaires les plus courants de Humira comprennent:
- Réactions du site d'injection: Rougettes qui gonflent des démangeaisons ou des ecchymoses. Ces symptômes disparaîtront généralement en quelques jours. Appelez votre médecin tout de suite si vous avez des rougeurs de douleur ou un gonflement autour du site d'injection qui ne disparaît pas en quelques jours ou empire.
- infections respiratoires supérieures (y compris sinus infections).
- mal de têtes.
- éruption cutanée.
Ce ne sont pas tous les effets secondaires possibles avec Humira. Dites à votre médecin si vous avez un effet secondaire qui vous dérange ou qui ne disparaît pas. Demandez à votre médecin ou à votre pharmacien plus d'informations.
Appelez votre médecin pour des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA à 1800- FDA-1088.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur à 36 ° F à 46 ° F (2 ° C à 8 ° C). Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas Freeze Humira. Ne pas Utilisez Humira si gelé même s'il a été décongelé.
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration imprimée sur le stylo à dose de carton Humira ou la seringue préreffilée. N'utilisez pas Humira après la date d'expiration.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) jusqu'à 14 jours. Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Jetez Humira s'il a été maintenu à température ambiante et n'a pas été utilisé dans les 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
- Ne pas use a Pen or prefilled syringe if the liquid is cloudy discolorouge or has flakes or particles in it.
- Ne pas drop or crush . The prefilled syringe is glass.
Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Informations générales sur l'utilisation sûre et efficace de Humira.
Les médicaments sont parfois prescrits à des fins autres que celles énumérées dans un guide de médicaments. N'utilisez pas Humira pour une condition pour laquelle il n'a pas été prescrit. Ne donnez pas Humira à d'autres personnes même si elles ont la même condition. Cela peut leur faire du mal. Ce guide de médicaments résume les informations les plus importantes sur Humira. Si vous souhaitez plus d'informations, parlez avec votre médecin. Vous pouvez demander à votre pharmacien ou médecin d'informations sur Humira qui est écrite pour les professionnels de la santé.
Quels sont les ingrédients de Humira?
Ingrédient actif: adalimumab
Pen 40 mg/0.8 mL 40 mg/0.8 mL prefilled syringe 20 mg/0.4 mL prefilled syringe 10 mg/0.2 mL prefilled syringe et 40 mg/0.8 mL institutional use vial:
Ingrédients inactifs: acide citrique monohydraté dibasique phosphate de sodium dihydraté mannitol monobasique phosphate de sodium dihydraté polydrate 80 chlorure de sodium citrate de sodium et eau pour l'injection. L'hydroxyde de sodium est ajouté si nécessaire pour régler le pH.
Pen 80 mg/0.8 mL 80 mg/0.8 mL prefilled syringe Pen 40 mg / 0,4 ml 40 mg / 0,4 ml prefilled syringe 20 mg/0.2 mL prefilled syringe et 10 mg/0.1 mL prefilled syringe:
Ingrédients inactifs: Mannitol Polysorbate 80 et eau pour l'injection.
Instructions pour une utilisation
®
(Hu-Mare-ah)
(adalimumab)
40 mg / 0,8 ml
Stylo à dose
Ne pas Essayez d'injecter vous-même Humira jusqu'à ce que vous ayez montré la bonne façon de donner les injections et d'avoir lu et comprendre ces instructions pour une utilisation. Si votre médecin décide que vous ou un soignant peut être en mesure de donner vos injections d'Humira à la maison, vous devriez recevoir une formation sur la bonne façon de préparer et d'injecter Humira. Il est important que vous lisiez comprendre et suivre ces instructions afin que vous injectez Humira de la bonne façon. Il est également important de parler à votre médecin pour être sûr de comprendre vos instructions de dosage Humira. Pour vous aider à vous rappeler quand injecter Humira, vous pouvez marquer votre calendrier à l'avance. Appelez votre fournisseur de soins de santé si vous ou votre soignant avez des questions sur la bonne façon d'injecter Humira.
IMPORTANT:
- Ne pas Utilisez Humira si gelé même s'il a été décongelé.
- Le stylo Humira contient du verre. Ne pas laisser tomber ou écraser le stylo car le verre à l'intérieur peut se casser.
- Chaque stylo Humira a 2 capuchons dessus. Ne retirez pas le capuchon gris (capuchon
- Lorsque le bouton de couleur prune sur le stylo Humira est enfoncé pour donner votre dose de Humira, vous entendrez un son de clic fort.
- Vous devez vous entraîner à injecter Humira avec votre médecin ou votre infirmière afin que vous ne soyez pas surpris par ce clic lorsque vous commencez à vous donner les injections à la maison.
- Le son de clic fort signifie le début de l'injection.
- Vous saurez que l'injection s'est terminée lorsque l'indicateur jaune apparaît complètement dans la vue de la fenêtre et cesse de se déplacer.
Voir la section ci-dessous appelée Préparer le stylo Humira.
Rassemblez les fournitures pour votre injection
- Vous aurez besoin des fournitures suivantes pour chaque injection de Humira.
Trouvez une surface plate propre pour placer les fournitures.
- 1 écouvillard d'alcool
- 1 boule de coton ou coussin de gaze (non inclus dans votre carton Humira)
- 1 stylo Humira (voir Figure A )
- Contasseur d'élimination des objets tranchants résistants à la crevaison pour l'élimination du stylo Humira (non inclus dans votre carton Humira). Voir le Comment dois-je jeter (éliminer) le stylo Humira utilisé? Section à la fin de ces instructions pour une utilisation.
Si plus à l'aise, sortez votre stylo Humira du réfrigérateur 15 à 30 minutes Avant d'injecter pour permettre au liquide d'atteindre la température ambiante. Ne pas Retirez le capuchon gris (capuchon Ne pas Humira chaud d'une autre manière (par exemple ne pas Réchauffez-le au micro-ondes ou dans de l'eau chaude).
Si vous n'avez pas toutes les fournitures dont vous avez besoin pour vous donner une injection, allez dans une pharmacie ou appelez votre pharmacien. La figure ci-dessous montre à quoi ressemble le stylo Humira. Voir Figure A .
Figure A
 |
Vérifiez le plateau de dose de carton et le stylo Humira.
Voir le Comment dois-je stocker Humira? Section à la fin de ces instructions pour une utilisation.
Figure B
 |
Figure C
 |
Choisissez le site d'injection
Préparer le site d'injection
Préparer le stylo Humira
Figure E
 |
Positionner le stylo et injecter Humira
Figure J
 |
- Assurez-vous que le nom Humira apparaît sur le plateau de dose de carton et l'étiquette du stylo Humira.
- Ne pas use et faire appel Votre médecin ou pharmacien si:
- Vous laissez tomber ou écrasez votre stylo Humira.
- Les joints en haut ou en bas du carton sont cassés ou manquants.
- La date d'expiration du plateau de dose de carton et du stylo est passée.
- Le stylo Humira a été gelé ou laissé en lumière directe du soleil.
- has been kept at room temperature for longer than 14 Les jours ou Humira ont été stockés au-dessus de 77 ° F (25 ° C).
- Tenez le stylo avec le capuchon gris (capuchon
- Assurez-vous que la quantité de liquide dans le stylo se trouve à la ligne de remplissage ou près de la ligne de remplissage vue à travers la fenêtre. C'est la pleine dose de Humira que vous injecterez. Voir Figure B .
- Si le stylo n'a pas la quantité totale de liquide ne pas use that Pen. Appelez votre pharmacien.
- Retournez le stylo et maintenez le stylo avec le capuchon gris (casquette Figure C .
- Vérifiez la solution à travers les fenêtres sur le côté du stylo pour vous assurer que le liquide est clair et incolore. Ne pas use Votre stylo Humira si le liquide est décoloré ou s'il contient des flocons ou des particules. Appelez votre pharmacien. Il est normal de voir une ou plusieurs bulles dans la fenêtre.
- Lavez et séchez bien vos mains.
- Choisissez un site d'injection sur:
Figure D

- le devant de vos cuisses ou
- votre bas de l'abdomen (ventre). Si vous choisissez votre abdomen, n'utilisez pas la zone de 2 pouces autour de votre nombril (nombril). Voir Figure D .
- Choisissez un autre site chaque fois que vous vous donnez une injection. Chaque nouvelle injection doit être donnée au moins un pouce d'un site que vous avez utilisé auparavant.
- Ne pas Injectez Humira dans la peau qui est:
- douloureux (tendre)
- meurtri
- rouge
- dur
- marqué ou où vous avez des vergetures
- Si vous avez du psoriasis ne pas Injectez directement dans toutes les plaques ou lésions cutanées rouges ou écailleuses surélevées sur votre peau.
- Ne pas inject through your clothes.
- Essuyez le site d'injection avec une préparation d'alcool (écouvillon) à l'aide d'un mouvement circulaire.
- Ne pas Touchez à nouveau cette zone avant de donner l'injection. Laissez la peau sécher avant l'injection. Ne pas ventilateur ou souffler sur la zone propre.
- Ne pas remove the gray cap (Cap # 1) or the plum-colorouge cap (Cap # 2) until right before your injection.
- Tenez le milieu du stylo (corps gris) avec une main afin que vous ne touchez pas le capuchon gris (capuchon Figure E .
- Avec votre autre main tirez le capuchon gris (capuchon Figure F .
- Jetez la casquette grise (casquette
Figure F
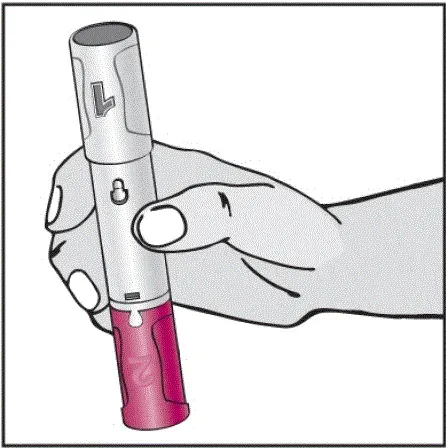
- Ne pas Mettez la casquette grise (casquette
- La manche à aiguille blanche qui couvre l'aiguille peut maintenant être vue.
- Ne pas Touchez l'aiguille avec vos doigts ou laissez l'aiguille toucher quoi que ce soit.
- Vous pouvez voir quelques gouttes de liquide sortir de l'aiguille. C'est normal.
- Retirez le capuchon de couleur prune (capuchon
Le bouton d'activateur de couleur prune:
Figure G

Figure H
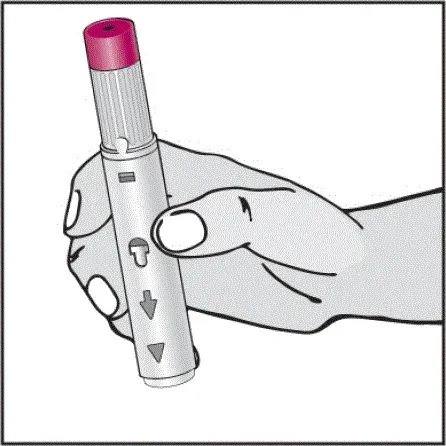
- Ne pas put the plum-colorouge cap (Cap # 2) back on le stylo because it could cause medicine to come out of the syringe.
- Tournez le stylo pour que le bouton d'activateur de couleur prune soit pointé vers le haut. Voir Figure G .
- Ne pas Appuyez sur le bouton Activateur de couleur prune jusqu'à ce que vous soyez prêt à injecter Humira. En appuyant sur le bouton Activateur de couleur prune libérera le médicament à partir du stylo.
- Tenez le stylo pour que vous puissiez voir la fenêtre. Voir Figure H . Il est normal de voir une ou plusieurs bulles dans la fenêtre.
- Positionner le stylo:
Figure I
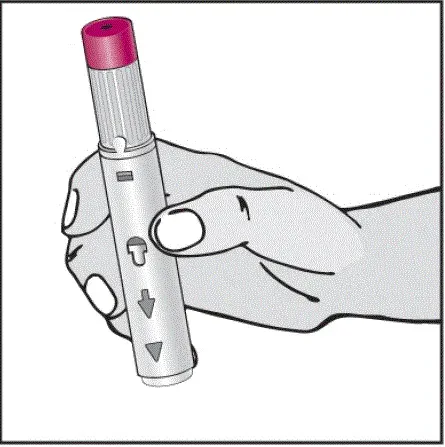
- Presser La zone de la peau nettoyée et maintenez-la fermement jusqu'à ce que l'injection soit terminée. Voir Figure I . Vous injecterez dans cette zone de peau surélevée.
- Placez l'extrémité blanche du stylo droit (à un angle de 90 °) et flat against the raised area of your skin that you are squeezing. Lieu le stylo so that it will pas inject the needle into your fingers that are holding the raised skin. Voir Figure J .
- Injecter Humira
Figure K

Figure L
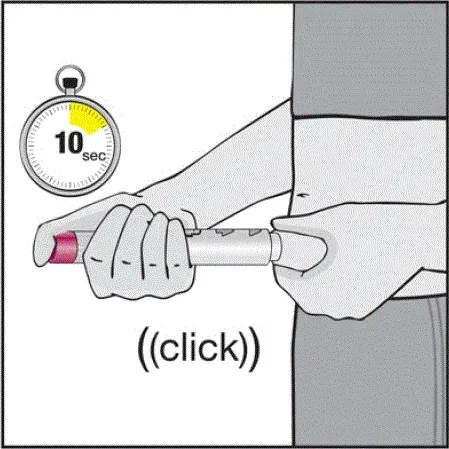
- Il est important que vous poussiez fermement le stylo vers le bas Tout au long du site d'injection avant de commencer l'injection.
- Continuez à pousser Pour empêcher le stylo de s'éloigner de la peau pendant l'injection.
- Appuyez sur le bouton Activateur de couleur prune avec votre pouce pour commencer l'injection. Essayez de ne pas couvrir la fenêtre. Voir Figure K .
- Vous entendrez un «clic» fort lorsque vous appuyez sur le bouton Activateur de couleur prune. Le clic fort signifie le début de l'injection.
- Continuez à appuyer sur le bouton Activateur de couleur prune et continuez à appuyer sur le stylo contre votre peau surélevée pressée jusqu'à ce que tous les médicaments soient injectés. Cela peut prendre jusqu'à 10 secondes, alors comptez lentement à dix. Continuez à pousser le stylo contre la peau surélevée pressée de votre site d'injection pendant tout le temps afin que vous obteniez la dose complète de médicaments.
- Vous saurez que l'injection s'est terminée lorsque l'indicateur jaune apparaît complètement dans la vue de la fenêtre et cesse de se déplacer. Voir Figure L .
- Lorsque l'injection est terminée, tirez lentement le stylo de votre peau. La manche à aiguille blanche se déplacera pour couvrir la pointe de l'aiguille. Voir Figure m .
Figure m
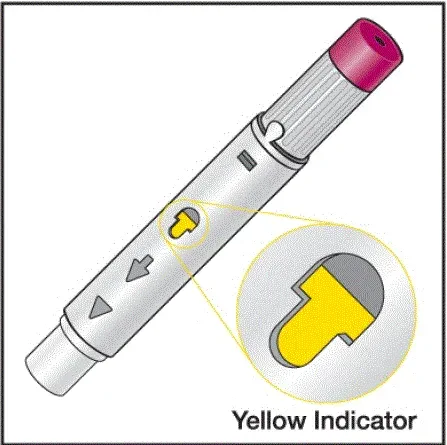
- Ne pas touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Il peut y avoir une petite quantité de liquide sur le site d'injection. C'est normal.
- Appuyez sur une boule de coton ou un coussin de gaze sur le site d'injection et maintenez-le pendant 10 secondes. Faire pas Frottez le site d'injection. Vous pouvez avoir un léger saignement. C'est normal.
- Jetez (disposer) votre stylo Humira usé dans un conteneur d'élimination des objets tranchants immédiatement après utilisation. Voir la section Comment devrais-je disposer du stylo Humira utilisé?
- Gardez un enregistrement des dates et de l'emplacement de vos sites d'injection. Pour vous aider à vous rappeler quand prendre Humira, vous pouvez marquer votre calendrier à l'avance.








Comment dois-je jeter (éliminer) le stylo Humira utilisé?
Figure N
 |
- Mettez immédiatement votre stylo dans un conteneur d'élimination des objets tranchants transportés par la FDA après utilisation. Voir Figure N . Ne pas throw away le stylo in your household téruption cutanée.
- Ne pas try to touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Si vous n'avez pas de conteneur d'élimination des objets tranchants approuvé par la FDA, vous pouvez utiliser un conteneur domestique qui est:
- fait d'un plastique robuste
- peut être fermé avec un couvercle résistant à la perforation serrée sans que les objets tranchants ne puissent sortir
- droit et stable pendant l'utilisation
- résistant à la fuite et
- correctement étiqueté pour avertir des déchets dangereux à l'intérieur du récipient.
- Lorsque votre conteneur d'élimination des objets tranchants est presque plein, vous devrez suivre vos directives communautaires pour la bonne façon de disposer de votre conteneur d'élimination des objets tranchants. Il peut y avoir des lois étatiques ou locales sur la façon dont vous devez jeter les aiguilles et les seringues d'occasion. Pour plus d'informations sur l'élimination des objets de reculations sûrs et pour des informations spécifiques sur l'élimination des objets tranchants dans l'État dans lequel vous vivez dans le site Web de la FDA à: https://www.fda.gov/safesharpsdisposal.
- Pour la sécurité et la santé de vous et d'autres ne réutilisez jamais vos stylos Humira.
- Les plateaux de dose et les emballages d'alcool d'alcool d'occasion peuvent être placés dans les déchets de votre ménage.
- Ne pas dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne pas recycle your used sharps disposal container.
- Gardez toujours le conteneur des objets tranchants hors de portée des enfants.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur entre 36 ° F et 46 ° F (2 ° C à 8 ° C). Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas Freeze Humira. Ne pas Utilisez Humira si gelé même s'il a été décongelé.
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration imprimée sur le bac à dose ou le stylo Humira Carton. Ne pas Utilisez Humira après la date d'expiration.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) pour jusqu'à 14 jours. Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Jetez humira s'il a été maintenu à température ambiante et n'a pas été utilisé à l'intérieur 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
- Ne pas use a Pen if the liquid is cloudy discolorouge or has flakes or particles in it.
- Ne pas drop or crush .
- Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Instructions pour une utilisation
®
(Hu-Mare-ah)
(adalimumab)
40 mg / 0,4 ml
Stylo à dose
Avant d'injecter: Votre fournisseur de soins de santé devrait vous montrer comment utiliser Humira avant de l'utiliser pour la première fois. Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) Si vous avez besoin d'aide.
Figure A: Stylo à dose
Système transdermique de fentanyl 100 mcg hr
 |
Informations importantes que vous devez savoir avant d'injecter Humira
Ne pas Utilisez le stylo et appelez votre fournisseur de soins de santé ou pharmacien si:
- Le liquide est décoloré décoloré ou contient des flocons ou des particules
- La date d'expiration est passée
- Le liquide a été gelé (même s'il est décongelé) ou laissé en lumière directe du soleil
- Le stylo a été abandonné ou écrasé
Gardez les capuchons jusqu'à juste avant votre injection.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur entre 36 ° F et 46 ° F (2 ° C à 8 ° C).
- Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas freeze
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration imprimée sur le bac à dose ou le stylo Humira Carton.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) pour jusqu'à 14 jours.
- Jetez Humira s'il a été maintenu à température ambiante et non utilisé à l'intérieur 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Lire des instructions sur toutes les pages avant d'utiliser le stylo Humira
Prendre out of the refrigerator.
Partir at room temperature for 15 à 30 minutes avant d'injecter.
- Ne pas Retirez le capuchon gris (capuchon
- Ne pas Humira chaud d'une autre manière. Par exemple ne pas Réchauffez-le au micro-ondes ou dans de l'eau chaude.
- Ne pas Utilisez le stylo si le liquide a été gelé (même s'il est décongelé)
 |
Vérifier Date d'expiration sur l'étiquette du stylo. Ne pas Utilisez le stylo si la date d'expiration est passée.
Lieu Ce qui suit sur une surface plate propre:
- 1 stylo à dose unique et écouvillon d'alcool
- 1 boule de coton ou coussin de gaze (non inclus)
- Conteneur d'élimination des objets tranchants résistants à la perforation (non inclus). Voir l'étape 9 à la fin de ces instructions pour une utilisation pour des instructions sur la façon de jeter (éliminer) votre stylo Humira
Se laver et sécher tes mains.
 |
Choisir Un site d'injection:
- Sur le devant de vos cuisses ou
- Votre abdomen (ventre) au moins 2 pouces de votre nombril (nombril)
- Différent de votre dernier site d'injection
Essuyer Le site d'injection dans un mouvement circulaire avec l'écouvillon d'alcool.
- Ne pas Injecter à travers les vêtements
- Ne pas Injecter dans la peau douloureuse et les cicatrices durs rouges ont des vergetures ou des zones avec des plaques de psoriasis
 |
Prise le stylo avec le capuchon gris Vérifier la fenêtre.
- Il est normal de voir 1 ou plusieurs bulles dans la fenêtre
- Assurez-vous que le liquide est clair et incolore
- Ne pas Utilisez le stylo si le liquide est décoloré ou a des flocons ou des particules
- Ne pas Utilisez le stylo s'il a été abandonné ou écrasé
 |
Tirer la casquette grise
Jetez le capuchon.
- Il est normal de voir quelques gouttes de liquide sortir de l'aiguille
Tirer la casquette de couleur prune
Jetez le capuchon.
Tournez le stylo de sorte que la flèche blanche pointe vers le site d'injection.
 |
Presser La peau de votre site d'injection pour faire une zone surélevée et le maintenir fermement jusqu'à ce que l'injection soit terminée.
Indiquer la flèche blanche vers le site d'injection.
Lieu la manche à aiguille blanche droite (Angle de 90 °) contre le site d'injection.
Prise le stylo afin que vous puissiez voir la fenêtre d'inspection.
Ne pas Appuyez sur le bouton Activateur Plum jusqu'à ce que vous soyez prêt à injecter.
 |
Il est important que vous poussiez fermement le stylo vers le bas Tout au long du site d'injection avant de commencer l'injection.
Continuez à pousser Pour empêcher le stylo de s'éloigner de la peau pendant l'injection.
Presse le bouton de l'activateur de prune et comptez lentement pour 10 secondes.
- Un clic fort signalera le commencer de l'injection
- Continuez à pousser le stylo fermement contre le site d'injection jusqu'à ce que l'injection soit terminée
- L'injection est terminée lorsque l'indicateur jaune a cessé de bouger
 |
Lorsque l'injection est terminée, tirez lentement le stylo de la peau. La manche à aiguille blanche couvrira la pointe de l'aiguille.
- Une petite quantité de liquide sur le site d'injection est normale
S'il y a plus de quelques gouttes de liquide sur l'appel du site d'injection 1-800-4HUMIRA (1-800448- 6472) pour l'aide.
Après avoir terminé l'injection, placez une boule de coton ou un coussin de gaze sur la peau du site d'injection.
- Ne pas frotter
- Un léger saignement au site d'injection est normal
 |
Comment devrais-je disposer du stylo Humira utilisé?
- Mettez immédiatement vos stylos d'aiguilles et les objets tranchants d'occasion dans un conteneur d'élimination des objets tranchants effacés de la FDA après utilisation. Ne pas throw away (dispose of) loose needles syringes et le stylo in the household téruption cutanée.
- Si vous n'avez pas de conteneur d'élimination des objets tranchants effacés de la FDA, vous pouvez utiliser un conteneur de ménage qui est:
- fait d'un plastique robuste
- peut être fermé avec un couvercle résistant à la perforation serrée sans que les objets tranchants ne puissent sortir
- droit et stable pendant l'utilisation
- résistant à la fuite et
- correctement étiqueté pour avertir des déchets dangereux à l'intérieur du récipient.
- Lorsque votre conteneur d'élimination des objets tranchants est presque plein, vous devrez suivre vos directives communautaires pour la bonne façon de disposer de votre conteneur d'élimination des objets tranchants. Il peut y avoir des lois étatiques ou locales sur la façon dont vous devez jeter les aiguilles et les seringues d'occasion. Pour plus d'informations sur l'élimination des objets tranchants sûrs et pour des informations spécifiques sur l'élimination des objets tranchants dans l'État dans lequel vous vivez sur le site Web de la FDA à: https://www.fda.gov/safesharpsdisposition.
- Ne pas dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne pas recycle your used sharps disposal container.
 |
Le stylo à stylos à bille en coton à l'écouvillon d'alcool ou à la dose de coton de gaze et de l'emballage peut être placé dans les déchets de votre ménage.
Questions sur l'utilisation du stylo Humira
Et si je n'ai pas reçu dans la formation en personne d'un fournisseur de soins de santé?
- Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) ou visitez www.humira.com si vous avez besoin d'aide
Comment savoir quand l'injection est terminée?
- L'indicateur jaune a cessé de bouger. Cela prend 10 secondes.
Que dois-je faire s'il y a plus de quelques gouttes de liquide sur le site d'injection?
- Appel 1-800-4HUMIRA (1-800-448-6472) pour obtenir de l'aide
Que se passe-t-il si je n'ai pas de conteneur d'élimination des objets tranchants approuvé par la FDA ou de conteneur de ménages approprié?
- Appel 1-800-4HUMIRA (1-800-448-6472) pour un conteneur d'élimination des objets tranchants en FDA gratuit
Toujours Gardez le stylo et le conteneur d'élimination des objets tranchants hors de portée des enfants.
Gardez une trace des dates et des emplacements de vos injections. Pour vous rappeler quand prendre Humira, marquez votre calendrier à l'avance.
 |
 |
Instructions pour une utilisation
®
(Hu-Mare-ah)
(adalimumab)
Packages contenant 80 mg / 0,8 ml
Stylo à dose
Avant d'injecter: Votre fournisseur de soins de santé devrait vous montrer comment utiliser Humira avant de l'utiliser pour la première fois. Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) Si vous avez besoin d'aide.
Figure A: Stylo à dose
 |
Informations importantes que vous devez savoir avant d'injecter Humira
Ne pas Utilisez le stylo et appelez votre fournisseur de soins de santé ou pharmacien si:
- Le liquide est décoloré décoloré ou contient des flocons ou des particules
- La date d'expiration est passée
- Le liquide a été gelé (même s'il est décongelé) ou laissé en lumière directe du soleil
- Le stylo a été abandonné ou écrasé
Gardez les capuchons jusqu'à juste avant votre injection.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur entre 36 ° F et 46 ° F (2 ° C à 8 ° C).
- Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas freeze
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration imprimée sur le bac à dose ou le stylo Humira Carton.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) pour jusqu'à 14 jours.
- Jetez Humira s'il a été maintenu à température ambiante et non utilisé à l'intérieur 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Lire des instructions sur toutes les pages avant d'utiliser le stylo Humira
Prendre out of the refrigerator.
Partir at room temperature for 15 à 30 minutes avant d'injecter.
- Ne pas Retirez le capuchon gris (capuchon
- Ne pas Humira chaud d'une autre manière. Par exemple ne pas le réchauffer au micro-ondes ou dans l'eau chaude
- Ne pas Utilisez le stylo si le liquide a été gelé (même s'il est décongelé)
 |
Vérifier Date d'expiration sur l'étiquette du stylo. Ne pas Utilisez le stylo si la date d'expiration est passée.
Lieu Ce qui suit sur une surface plate propre:
- 1 stylo à dose unique et écouvillon d'alcool
- 1 boule de coton ou coussin de gaze (non inclus)
- Conteneur d'élimination des objets tranchants résistants à la perforation (non inclus). Voir l'étape 9 à la fin de ces instructions pour une utilisation pour des instructions sur la façon de jeter (éliminer) votre stylo Humira
Se laver et sécher tes mains.
 |
Choisir Un site d'injection:
- Sur le devant de vos cuisses ou
- Votre abdomen (ventre) au moins 2 pouces de votre nombril (nombril)
- Différent de votre dernier site d'injection
Essuyer Le site d'injection dans un mouvement circulaire avec l'écouvillon d'alcool.
- Ne pas Injecter à travers les vêtements
- Ne pas Injecter dans la peau douloureuse et les cicatrices durs rouges ont des vergetures ou des zones avec des plaques de psoriasis
 |
Prise le stylo avec le capuchon gris Vérifier la fenêtre.
- Il est normal de voir 1 ou plusieurs bulles dans la fenêtre
- Assurez-vous que le liquide est clair et incolore
- Ne pas Utilisez le stylo si le liquide est décoloré ou a des flocons ou des particules
- Ne pas Utilisez le stylo s'il a été abandonné ou écrasé
 |
Tirer la casquette grise
Jetez le capuchon.
- Il est normal de voir quelques gouttes de liquide sortir de l'aiguille
Tirer la casquette de couleur prune
Jetez le capuchon.
Tourner le stylo so that the white arrow points toward the injection site.
 |
Presser La peau de votre site d'injection pour faire une zone surélevée et le maintenir fermement jusqu'à ce que l'injection soit terminée.
Indiquer la flèche blanche vers le site d'injection.
Lieu la manche à aiguille blanche droite (Angle de 90 °) contre le site d'injection.
Prise le stylo afin que vous puissiez voir la fenêtre d'inspection.
 |
Ne pas Appuyez sur le bouton Activateur Plum jusqu'à ce que vous soyez prêt à injecter.
Il est important que vous poussiez fermement le stylo vers le bas Tout au long du site d'injection avant de commencer l'injection.
Presse le bouton de l'activateur de prune et comptez lentement pour 15 secondes.
- Un clic fort signalera le commencer de l'injection
- Continuez à pousser le stylo fermement contre le site d'injection jusqu'à ce que l'injection soit terminée
- L'injection est terminée lorsque l'indicateur jaune a cessé de bouger
 |
Lorsque l'injection est terminée, tirez lentement le stylo de la peau. La manche à aiguille blanche couvrira la pointe de l'aiguille.
- Une petite quantité de liquide sur le site d'injection est normale
S'il y a plus de quelques gouttes de liquide sur l'appel du site d'injection 1-800-4HUMIRA (1-800448- 6472) pour l'aide.
Après avoir terminé l'injection, placez une boule de coton ou un coussin de gaze sur la peau du site d'injection.
- Ne pas frotter
- Un léger saignement au site d'injection est normal
 |
Comment devrais-je disposer du stylo Humira utilisé?
- Mettez immédiatement vos stylos d'aiguilles et les objets tranchants d'occasion dans un conteneur d'élimination des objets tranchants effacés de la FDA après utilisation. Ne pas throw away (dispose of) loose needles syringes et le stylo in the household téruption cutanée.
- Si vous n'avez pas de conteneur d'élimination des objets tranchants approuvé par la FDA, vous pouvez utiliser un conteneur domestique qui est:
- fait d'un plastique robuste
- peut être fermé avec un couvercle résistant à la perforation serrée sans que les objets tranchants ne puissent sortir
- droit et stable pendant l'utilisation
- résistant à la fuite et
- correctement étiqueté pour avertir des déchets dangereux à l'intérieur du récipient.
- Lorsque votre conteneur d'élimination des objets tranchants est presque plein, vous devrez suivre vos directives communautaires pour la bonne façon de disposer de votre conteneur d'élimination des objets tranchants. Il peut y avoir des lois étatiques ou locales sur la façon dont vous devez jeter les aiguilles et les seringues d'occasion. Pour plus d'informations sur l'élimination des objets tranchants sûrs et pour des informations spécifiques sur l'élimination des objets tranchants dans l'État dans lequel vous vivez sur le site Web de la FDA à: https://www.fda.gov/safesharpsdisposition.
- Ne pas dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne pas recycle your used sharps disposal container.
 |
Le stylo à stylos à bille en coton à l'écouvillon d'alcool ou à la dose de coton de gaze et de l'emballage peut être placé dans les déchets de votre ménage.
Questions sur l'utilisation du stylo Humira
Et si je n'ai pas reçu de formation en personne d'un fournisseur de soins de santé?
- Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) ou visitez www.humira.com si vous avez besoin d'aide
Comment savoir quand l'injection est terminée?
- L'indicateur jaune a cessé de bouger. Cela prend 15 secondes.
Que dois-je faire s'il y a plus de quelques gouttes de liquide sur le site d'injection?
- Appel 1-800-4HUMIRA (1-800-448-6472) pour obtenir de l'aide
Que se passe-t-il si je n'ai pas de conteneur d'élimination des objets tranchants approuvé par la FDA ou de conteneur de ménages approprié?
- Appel 1-800-4HUMIRA (1-800-448-6472) pour un conteneur d'élimination des objets tranchants en FDA gratuit
Toujours Gardez le stylo et le conteneur d'élimination des objets tranchants hors de portée des enfants.
Gardez une trace des dates et des emplacements de vos injections. Pour vous rappeler quand prendre Humira, marquez votre calendrier à l'avance.
 |
 |
Instructions pour une utilisation
®
(Hu-Mare-ah)
(adalimumab)
40 mg / 0,8 ml 20 MG/0.4 ML AND 10 MG/0.2 ML
Seringue pré-remplie à dose
Ne pas Essayez d'injecter vous-même Humira jusqu'à ce que vous ayez montré la bonne façon de donner les injections et d'avoir lu et comprendre ces instructions pour une utilisation. Si votre médecin décide que vous ou un soignant peut être en mesure de donner vos injections d'Humira à la maison, vous devriez recevoir une formation sur la bonne façon de préparer et d'injecter Humira. Il est important que vous lisiez comprendre et suivre ces instructions afin que vous injectez Humira de la bonne façon. Il est également important de parler à votre médecin pour être sûr de comprendre vos instructions de dosage Humira. Pour vous aider à vous rappeler quand injecter Humira, vous pouvez marquer votre calendrier à l'avance. Appelez votre fournisseur de soins de santé si vous ou votre soignant avez des questions sur la bonne façon d'injecter Humira.
Rassemblez les fournitures pour votre injection
- Vous aurez besoin des fournitures suivantes pour chaque injection de Humira.
Trouvez une surface plate propre pour placer les fournitures.
- 1 écouvillard d'alcool
- 1 boule de coton ou coussin de gaze (non inclus dans votre carton Humira)
- 1 seringue préreffilée Humira (voir Figure A )
- Rétinément d'élimination des objets tranchants résistants à la crevaison pour l'élimination de la seringue préreadis Humira (non incluse dans votre Carton Humira). Voir le Comment dois-je jeter (éliminer) les seringues et les aiguilles préreffies utilisées? Section à la fin de ces instructions pour une utilisation.
Si plus à l'aise, prenez votre seringue préreffilée Humira hors du réfrigérateur 15 à 30 minutes Avant d'injecter pour permettre au liquide d'atteindre la température ambiante. Ne pas Retirez le couvercle de l'aiguille tout en lui permettant d'atteindre la température ambiante. Ne pas Humira chaud d'une autre manière (par exemple ne pas Réchauffez-le au micro-ondes ou dans de l'eau chaude).
Si vous n'avez pas toutes les fournitures dont vous avez besoin pour vous donner une injection, allez dans une pharmacie ou appelez votre pharmacien.
La figure ci-dessous montre à quoi ressemble une seringue prérempli. Voir Figure A .
Figure A
 |
Vérifier the carton dose tray et prefilled syringe
Voir le Comment dois-je stocker Humira? Section à la fin de ces instructions pour une utilisation.
Choisissez le site d'injection
Préparer le site d'injection
Ne pas ventilateur ou souffler sur la zone propre.
Préparez la seringue et l'aiguille
Figure D
 |
Positionner la seringue préreffilée et injecter Humira
Positionner la seringue
Figure H
 |
Injecter Humira
- Assurez-vous que le nom Humira apparaît sur le plateau de dose et l'étiquette de seringue prérempli.
- Ne pas use et faire appel Votre médecin ou pharmacien si:
- Les joints en haut ou en bas du carton sont cassés ou manquants.
- L'étiquetage Humira a une date expirée. Vérifiez la date d'expiration sur votre carton Humira et ne pas Utilisez si la date est passée.
- La seringue pré-remplie a été gelée ou laissée en lumière directe du soleil.
- has been kept at room temperature for longer than 14 Les jours ou Humira ont été stockés au-dessus de 77 ° F (25 ° C).
- Le liquide dans la seringue prérempli est décoloré ou contient des flocons ou des particules. Assurez-vous que le liquide est clair et incolore.
- Lavez et séchez bien vos mains.
- Choisissez un site d'injection sur:
Figure B
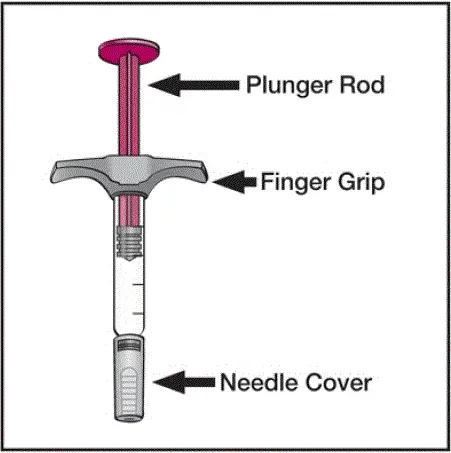
- le devant de vos cuisses ou
- votre bas de l'abdomen (ventre). Si vous choisissez votre abdomen, n'utilisez pas la zone de 2 pouces autour de votre nombril (nombril). Voir Figure B .
- Choisissez un autre site chaque fois que vous vous donnez une injection. Chaque nouvelle injection doit être donnée au moins un pouce d'un site que vous avez utilisé auparavant.
- Ne pas Injecter dans la peau qui est:
- douloureux (tendre)
- meurtri
- rouge
- dur
- marqué ou où vous avez des vergetures
- Si vous avez du psoriasis ne pas Injectez directement dans toutes les plaques ou lésions cutanées rouges ou écailleuses surélevées sur votre peau.
- Ne pas inject through your clothes.
- Essuyez le site d'injection avec une préparation d'alcool (écouvillon) à l'aide d'un mouvement circulaire.
- Faire pas Touchez à nouveau cette zone avant de donner l'injection. Laissez la peau sécher avant l'injection.
- Vérifier the fluid level in the syringe:
Figure C
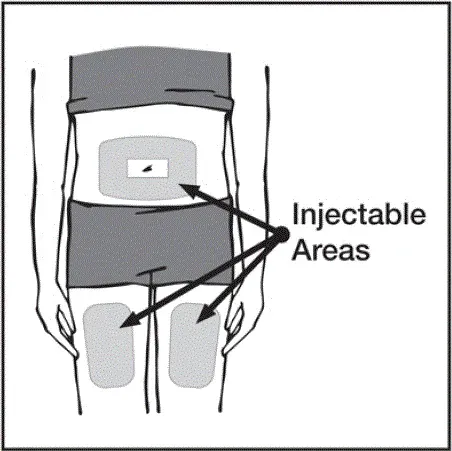
- Prise the syringe with the coverouge needle pointing down. Voir Figure C .
- Prise the syringe at eye level. Look closely to make sure that the amount of liquid in the syringe is the same or close to the:
- Ligne de 0,8 ml pour la seringue pré-remplie de 40 mg. Voir Figure D .
- Ligne de 0,4 ml pour la seringue pré-remplie de 20 mg. Voir Figure D .
- Ligne de 0,2 ml pour la seringue pré-remplie de 10 mg. Voir Figure D .
- Le haut du liquide peut être incurvé. Si la seringue n'a pas la quantité correcte de liquide ne pas use that syringe. Appelez votre pharmacien.
- Retirez le couvercle de l'aiguille:
Figure E
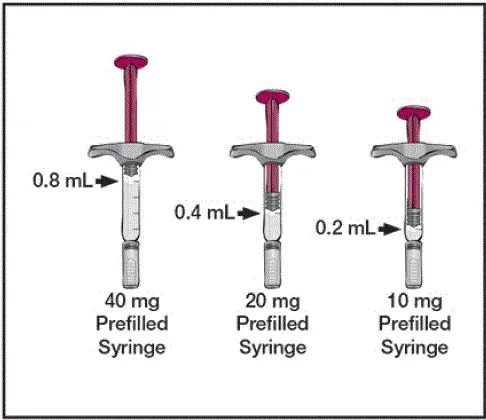
- Prise the syringe in one het. With the other het gently remove the needle cover. Voir Figure E .
- Jetez la couverture d'aiguille.
- Ne pas Touchez l'aiguille avec vos doigts ou laissez l'aiguille toucher quoi que ce soit.
- Tourner the syringe so the needle is facing up et hold the syringe at eye level with one het so you can see the air in the syringe. Utiliser votre autre main pousser lentement le piston pour pousser l'air à travers l'aiguille. Voir Figure F .
Figure F
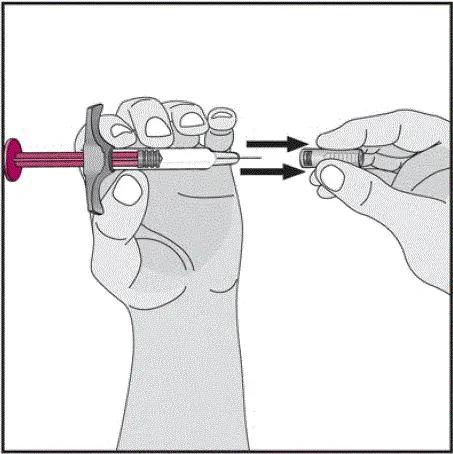
- Vous pouvez voir une goutte de liquide au bout de l'aiguille. C'est normal.
- Prise the body of the prefilled syringe in one het between the thumb et index finger. Prise the syringe in your het like a pencil. Voir Figure G .
Figure G
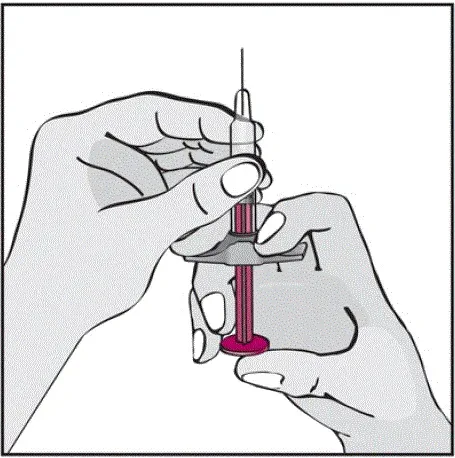
- Ne pas Retirez le piston à tout moment.
- Avec votre autre main, pressez doucement la zone de la peau nettoyée et maintenez-la fermement. Voir Figure H .
- En utilisant un mouvement rapide en forme de fléchette, insérez l'aiguille dans la peau pressée à environ un Angle de 45 degrés. Voir Figure I .
Figure I
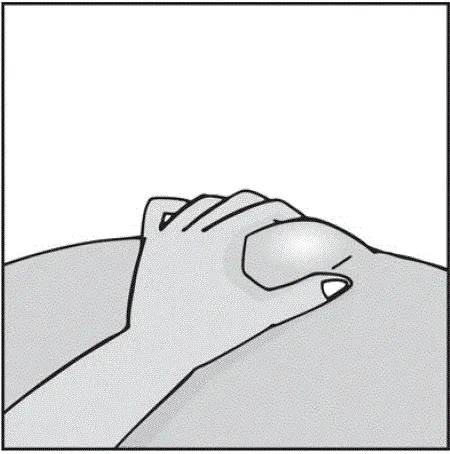
- Une fois que l'aiguille est en lâchement de la peau. Retirez doucement sur le piston.
Si le sang apparaît dans la seringue:
Figure J
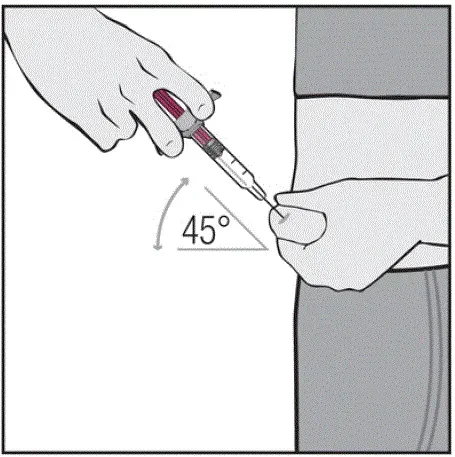
- Cela signifie que vous êtes entré dans un vaisseau sanguin.
- Ne pas inject .
- Tirer the needle out of the skin while keeping the syringe at the same angle.
- Presse a cotton ball or gauze pad over the injection site et hold it for 10 secondes. Voir Figure J .
- Ne pas Utilisez à nouveau la même seringue et aiguille. Jetez l'aiguille et la seringue dans votre conteneur des objets tranchants.
- Ne pas Frottez le site d'injection. Vous pouvez avoir un léger saignement. C'est normal.
- Répétez les étapes 1 à 12 avec une nouvelle seringue préreffilée.
Si aucun sang n'apparaît dans la seringue:
- Poussez lentement le piston jusqu'à ce que tout le liquide soit injecté et que la seringue soit vide.
- Tirer the needle out of the skin while keeping the syringe at the same angle.
- Appuyez sur une boule de coton ou un coussin de gaze sur le site d'injection et maintenez-le pendant 10 secondes. Faire pas Frottez le site d'injection. Vous pouvez avoir un léger saignement. C'est normal.







- Jetez la seringue et l'aiguille préremplies d'occasion dans un conteneur d'élimination des objets tranchants immédiatement après utilisation. Voir Comment dois-je jeter (éliminer) des seringues et des aiguilles préreffies utilisées?
- Gardez un enregistrement des dates et de l'emplacement de vos sites d'injection. Pour vous aider à vous rappeler quand prendre Humira, vous pouvez marquer votre calendrier à l'avance.
Comment dois-je jeter (éliminer) des seringues et des aiguilles préreffies utilisées?
Figure K
 |
- Mettez immédiatement vos aiguilles et seringues d'occasion dans un conteneur d'élimination des objets tranchants transportés par la FDA après utilisation. Voir Figure K . Ne pas throw away (dispose of) loose needles et syringes in your household téruption cutanée.
- Ne pas try to touch the needle.
- Si vous n'avez pas de conteneur d'élimination des objets tranchants approuvé par la FDA, vous pouvez utiliser un conteneur domestique qui est:
- fait d'un plastique robuste
- peut être fermé avec un couvercle résistant à la perforation serrée sans que les objets tranchants ne puissent sortir
- droit et stable pendant l'utilisation
- résistant à la fuite et
- correctement étiqueté pour avertir des déchets dangereux à l'intérieur du récipient.
- Lorsque votre conteneur d'élimination des objets tranchants est presque plein, vous devrez suivre vos directives communautaires pour la bonne façon de disposer de votre conteneur d'élimination des objets tranchants. Il peut y avoir des lois étatiques ou locales sur la façon dont vous devez jeter les aiguilles et les seringues d'occasion. Pour plus d'informations sur l'élimination des objets de reculations sûrs et pour des informations spécifiques sur l'élimination des objets tranchants dans l'État dans lequel vous vivez dans le site Web de la FDA à: https://www.fda.gov/safesharpsdisposal.
- Pour la sécurité et la santé de vous et d'autres aiguilles et des seringues utilisées ne doivent jamais être réutilisées.
- Les plateaux de dose et les emballages d'alcool d'alcool d'occasion peuvent être placés dans les déchets de votre ménage.
- Ne pas dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne pas recycle your used sharps disposal container.
- Gardez toujours le conteneur des objets tranchants hors de portée des enfants.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur entre 36 ° F et 46 ° F (2 ° C à 8 ° C). Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas Freeze Humira. Ne pas Utilisez Humira si gelé même s'il a été décongelé.
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration soit imprimée sur le plateau de dose de carton Humira ou la seringue préreffilée. Ne pas Utilisez Humira après la date d'expiration.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) pour jusqu'à 14 jours. Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Jetez humira s'il a été maintenu à température ambiante et n'a pas été utilisé à l'intérieur 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
- Ne pas use a prefilled syringe if the liquid is cloudy discolorouge or has flakes or particles in it.
- Ne pas drop or crush . The prefilled syringe is glass.
- Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Instructions pour une utilisation
®
(Hu-Mare-ah)
(adalimumab)
80 mg / 0,8 ml 40 mg / 0,4 ml 20 mg / 0,2 ml et 10 mg / 0,1 ml
Seringue pré-remplie à dose
Avant d'injecter: Votre fournisseur de soins de santé devrait vous montrer comment utiliser Humira avant de l'utiliser pour la première fois. Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) Si vous avez besoin d'aide.
Figure A: Single-Fairese Prefille
 |
Informations importantes que vous devez savoir avant d'injecter Humira
Ne pas Utilisez la seringue préférée et appelez votre fournisseur de soins de santé ou votre pharmacien si:
- Le liquide est décoloré décoloré ou contient des flocons ou des particules
- La date d'expiration est passée
- Le liquide a été gelé (même s'il est décongelé) ou laissé en lumière directe du soleil
- La seringue préremplie a été abandonnée ou écrasée
Gardez la couverture à l'aiguille jusqu'à juste avant votre injection.
Comment dois-je stocker Humira?
- Conservez Humira dans le réfrigérateur entre 36 ° F et 46 ° F (2 ° C à 8 ° C).
- Stockez Humira dans le carton d'origine jusqu'à utiliser pour le protéger de la lumière.
- Ne pas freeze
- Humira réfrigéré peut être utilisé jusqu'à ce que la date d'expiration soit imprimée sur le plateau de dose de carton Humira ou la seringue préreffilée.
- Si nécessaire par exemple lorsque vous voyagez, vous pouvez également stocker Humira à température ambiante jusqu'à 77 ° F (25 ° C) pour jusqu'à 14 jours.
- Jetez Humira s'il a été maintenu à température ambiante et non utilisé à l'intérieur 14 jours.
- Enregistrez la date à laquelle vous retirez d'abord Humira du réfrigérateur dans les espaces fournis sur le carton et le bac à dose.
- Ne stockez pas Humira dans une chaleur ou un froid extrême.
Gardez les fournitures d'injection Humira et tous les autres médicaments hors de portée des enfants.
Lire des instructions sur toutes les pages avant d'utiliser la seringue préremphed à dose Humira
Prendre out of the refrigerator.
Partir at room temperature for 15 à 30 minutes avant d'injecter.
- Ne pas Retirez le couvercle de l'aiguille tout en permettant à Humira d'atteindre la température ambiante
- Ne pas Humira chaud d'une autre manière. Par exemple ne pas Réchauffez-le au micro-ondes ou dans de l'eau chaude.
- Ne pas Utilisez la seringue prérempillée si le liquide a été congelé (même s'il est décongelé)
 |
Vérifier Date d'expiration sur l'étiquette de seringue prérempli. Ne pas Utilisez la seringue pré-remplie si la date d'expiration est passée.
Lieu Ce qui suit sur une surface plate propre:
- 1 seringue pré-remplie à dose et écouvillon d'alcool
- 1 boule de coton ou coussin de gaze (non inclus)
- Conteneur d'élimination des objets tranchants résistants à la perforation (non inclus). Voir l'étape 9 à la fin de ces instructions pour une utilisation pour des instructions sur la façon de jeter (éliminer) votre seringue prérempli
Se laver et sécher tes mains.
 |
Choisir Un site d'injection:
- Sur le devant de vos cuisses ou
- Votre abdomen (ventre) au moins 2 pouces de votre nombril (nombril)
- Différent de votre dernier site d'injection
Essuyer Le site d'injection dans un mouvement circulaire avec l'écouvillon d'alcool.
- Ne pas Injecter à travers les vêtements
- Ne pas Injecter dans la peau douloureuse et les cicatrices durs rouges ont des vergetures ou des zones avec des plaques de psoriasis
 |
Prise la seringue prérempliée dans une main.
Tirer doucement L'aiguille couvre directement avec l'autre main.
- Jeter la couverture à l'aiguille
- Ne pas Touchez l'aiguille avec vos doigts ou laissez l'aiguille toucher quoi que ce soit
 |
Prise La seringue prérempliée avec l'aiguille tournée vers le haut.
- Prise La seringue prérempliée au niveau des yeux avec une main afin que vous puissiez voir l'air dans la seringue préreffilée
- Utiliser votre autre main pousser lentement le piston pour pousser l'air à travers l'aiguille.
- Vous pouvez voir une goutte de liquide au bout de l'aiguille. C'est normal.
 |
Prise Le corps de la seringue pré-remplie dans une main entre le pouce et les doigts d'index. Tenez la seringue préférée dans votre main comme un crayon.
Ne pas Retirez le piston à tout moment.
 |
Presser doucement La zone de la peau nettoyée sur votre site d'injection avec votre autre main. Tenez fermement la peau.
Insérer L'aiguille dans la peau à environ un angle de 45 degrés en utilisant un mouvement rapide de type fléchette.
- Une fois que l'aiguille est en lâchement de la peau.
Pousser lentement Le piston jusqu'à ce que tout le liquide soit injecté et que la seringue pré-remplie soit vide.
 |
Lorsque l'injection est terminée, tirez lentement l'aiguille hors de la peau tout en gardant la seringue prérempillée à l'angle.
Après avoir terminé l'injection, placez une boule de coton ou un coussin de gaze sur la peau du site d'injection.
- Ne pas frotter
- Un léger saignement au site d'injection est normal
 |
Comment devrais-je éliminer la seringue préreffilée utilisée Humira?
- Mettez immédiatement vos seringues d'aiguilles et les objets tranchants usagés dans un conteneur d'élimination des objets tranchants approuvé par la FDA après utilisation. Ne pas throw away (dispose of) loose needles et syringes in the household téruption cutanée.
- Si vous n'avez pas de conteneur d'élimination des objets tranchants approuvé par la FDA, vous pouvez utiliser un conteneur domestique qui est:
- fait d'un plastique robuste
- peut être fermé avec un couvercle résistant à la perforation serrée sans que les objets tranchants ne puissent sortir
- droit et stable pendant l'utilisation
- résistant à la fuite et
- correctement étiqueté pour avertir des déchets dangereux à l'intérieur du récipient.
- Lorsque votre conteneur d'élimination des objets tranchants est presque plein, vous devrez suivre vos directives communautaires pour la bonne façon de disposer de votre conteneur d'élimination des objets tranchants. Il peut y avoir des lois étatiques ou locales sur la façon dont vous devez jeter les aiguilles et les seringues d'occasion. Pour plus d'informations sur l'élimination des objets tranchants sûrs et pour des informations spécifiques sur l'élimination des objets tranchants dans l'État dans lequel vous vivez sur le site Web de la FDA à: https://www.fda.gov/safesharpsdisposition.
- Ne pas dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne pas recycle your used sharps disposal container.
 |
La boule de coton ou un plateau de dose de coton à coton ou un coton de gaze et l'emballage peuvent être placés dans vos déchets domestiques.
Questions sur l'utilisation de la seringue pré-remplie à dose Humira
Et si je n'ai pas reçu de formation en personne d'un fournisseur de soins de santé?
- Appelez votre fournisseur de soins de santé ou 1-800-4HUMIRA (1-800-448-6472) ou visitez www.humira.com si vous avez besoin d'aide
Que se passe-t-il si je n'ai pas de conteneur d'élimination des objets tranchants approuvé par la FDA ou de conteneur de ménages approprié?
- Appel 1-800-4HUMIRA (1-800-448-6472) pour un conteneur d'élimination des objets tranchants en FDA gratuit
Toujours Gardez la seringue prérempli et le conteneur d'élimination des objets tranchants hors de portée des enfants.
Gardez une trace des dates et des emplacements de vos injections. Pour vous rappeler quand prendre Humira, marquez votre calendrier à l'avance.
 |
 |
Ces instructions pour une utilisation ont été approuvées par la Food and Drug Administration des États-Unis.